Chapter 3
Pharmacokinetics
3.1 The scope of pharmacokinetics
Pharmacokinetics deals with the following questions:
- Will the drug reach its intended site of action? If not, can we improve the drug’s uptake and distribution to help it reach its target?
- After uptake, how long will the drug stay in the system? How is it eliminated from the system?
As we will see, getting a drugs to its target can be a considerable challenge. Many experimental drugs that are viable in principle and can be shown to work in vitro are not useful in vivo because of this difficulty.
And no, the latest and greatest nanoparticles most likely aren’t going to be the answer to this problem. You may have noticed that most people who flog their revolutionary nanoparticle contraptions for “drug delivery” don’t even bother mentioning any specific drugs that they are going to deliver. This should tell us something.
3.2 Stages of drug transport
- Absorption: Uptake of the drug from the compartment of application into the blood plasma
- Distribution: Equilibration of the drug between the blood plasma and the rest of the organism
- Elimination: Excretion or metabolic inactivation of the drug
Notes: These stages do not occur strictly successively but overlap in time: As soon as some drug molecules have been taken up, they will start to distribute and undergo elimination, even while most others are still waiting for uptake. Moreover, strictly speaking, equilibrium of distribution can be reached only when a drug is administered continuously, e.g. by intravenous infusion, which is not usually the case. With this out of the way, we will now consider these stages separately and in turn.
3.2.1 Absorption depends on the route of application
| Route | Advantage | Disadvantage |
| Oral | Convenience—route of choice where possible | Multiple barriers between intestine and circulation |
| Intravenous | No barriers to absorption | Involved; risk of infection; allergic reactions more severe |
| Pulmonary | Fast, quantitative uptake | Limited to gases (mostly narcotics) |
Notes: Oral application of drugs is the most common case, because it is the most convenient for the patients. However, the obstacles to uptake from the gut into the circulation are formidable; we have already seen examples of drugs—remikiren and saralasin, see slide 1.2.6f—that fail entirely to be absorbed after oral application.
The second-most common route of application is intravenous injection or infusion. In this case, the absorption stage is bypassed altogether. This route is used with most protein drugs, as well as with small molecules that fail to be taken up after oral ingestion.
In clinical emergencies, intravenous application is usually preferred even with drugs that are suitable for oral application in principle, in order to ensure their rapid and quantitative uptake.
3.2.2 Distribution depends on the location of the drug target
| Location of target | Example |
| Inside circulation, outside cells | Proteases in blood coagulation and fibrinolysis |
| Inside circulation, inside cells | Chemotherapy of malaria parasites |
| Outside circulation, on cell surfaces | Histamine receptors |
| Outside circulation, inside cells | Cyclooxygenase |
Notes: After reaching the circulation, a drug may have to cross fewer or more barriers along the way to its target. In the most favorable case, the targets are located right within the circulation, and not hidden within any cells. This applies to the proteases involved in blood clot formation and dissolution (fibrinolysis), as well as to receptors that are located on the surfaces of thrombocytes or leukocytes.
3.3 Drug transport across anatomical barriers
Anatomical barriers concern all stages of drug transport. They differ in their active and passive transport properties. Because of its central place in drug transport, the blood circulation and its anatomical properties are particularly pertinent.
3.3.1 Sections of the blood circulation
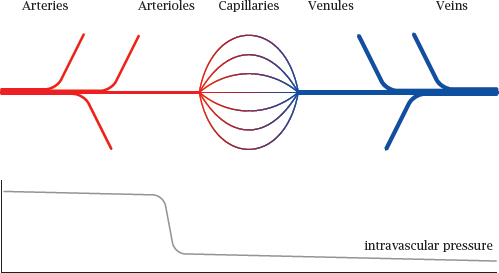
Notes: In the general circulation, oxygen-rich blood flows in the arteries. The final branches of the arteries are the arterioles. These vessels have the greatest flow resistance, and therefore the intravascular blood pressure, which is high in the arteries, here drops steeply to the much lower level that prevails in the capillaries and veins.
Both the arteries and veins have walls with multiple continuous layers of tissue that have low permeability for gases and metabolites. The exchange of solutes between blood and tissues is mostly confined to the capillaries.
3.3.2 Capillaries as barriers to drug transport
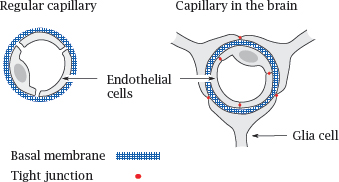
In almost all tissues outside the brain, the capillary walls have a loose, leaky structure, such that there are gaps between, and fenestrations (windows) across the endothelial cells. The only continuous structure that spans these gaps and fenestrations is the basal membrane, which therefore forms the only barrier to substance exchange by diffusion. It acts like a dialysis membrane; molecules smaller than 10 kDa can pass it freely, whereas blood plasma proteins and other macromolecules are retained.
In the brain and the spinal cord, the endothelia of the capillaries don’t have fenestrations, and adjacent cells are connected by tight junctions. On the outer side, the basal membrane of each capillary is surrounded by another tightly sealed cellular layer, which is formed by glia cells. Therefore, instead of a single, porous basal membrane, the lumen of the capillary is separated from the surrounding tissue by two continuous cell layers. This structure, which is much less permissive toward substance exchange than an ordinary capillary wall, forms the blood brain barrier.
The stacked cell membranes of the blood brain barrier prevent many small molecules from entering the central nervous system. In addition to passively impeding diffusion, the endothelial cells also contain ABC transporters that actively eject many drug molecules from the cytosol right back into the circulation. These transporters further reduce the penetration of drugs into the brain (see slide 3.5.3).
3.3.3 The intestinal epithelium as a barrier to drug absorption
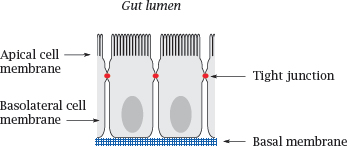
Notes: The small intestine is the place in which most orally ingested drugs are taken up. The epithelial cells sit atop a basal membrane and are joined to each other by tight junctions; therefore, drug molecules (and nutrients) have to be taken up across the cell membranes.
Like the endothelial cells in the blood brain barrier, the intestinal epithelia also express ABC transporters, which extrude some solutes from the cells back into the gut lumen. In addition, these cells also express cytochrome P450 enzymes, which metabolize and inactivate many drug molecules. The role of cytochrome P450 enzymes in drug metabolism is covered in detail in Chapter 4.
3.4 Drug transport across cell membranes
As we have seen, cell membranes restrict drug transport across the intestine and into the brain. In addition, they will also restrict the cellular uptake of drugs that act on intracellular targets.
3.4.1 Mechanisms of solute transport across cell membranes
- Active transport
1. Primary: ATP-coupled
2. Secondary: driven by ion gradients
- Passive transport
3. Facilitated diffusion: protein-mediated transport, not coupled to ATP or ion gradients
4. Non-facilitated diffusion of lipophilic compounds, non-ionic diffusion
Notes: Transport of types 1-3 is mediated by transport proteins and is substrate-specific. Most drugs don’t fit the substrate specificities of any of these transporters, so they have to cross cell membranes by non-facilitated diffusion, which occurs directly across the lipid bilayer and is independent of membrane proteins. The efficiency of this mode of transport depends mostly on the drug’s physicochemical properties.
3.4.2 Structure of a phosphatidylcholine bilayer
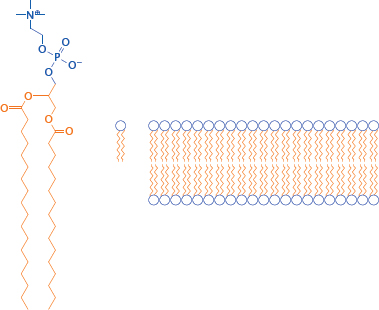
Notes: Obviously, cell membranes are more complex than a simple phosphatidylcholine (PC) bilayer; about 50% of a cell membrane consists of various membrane proteins, and the remainder is composed of a fairly complex mixture of lipid molecules. Nevertheless, a simple PC membrane is a useful model for drug transport across cell membranes by passive diffusion.
The solvent interfaces of a PC bilayer consist of the hydrophilic glycerophosphocholine headgroups, whereas the core consists of the aggregated fatty acyl side chains.
3.4.3 The polarity of drug molecules affects their rate of diffusion across lipid bilayers
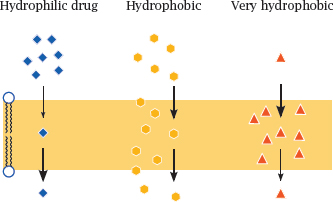
Notes: The ability of drug molecules to cross a lipid bilayer by passive diffusion correlates with their ability to partition into and out of the hydrophobic membrane interior. A fairly but not excessively hydrophobic character is most conducive to transport; very polar molecules will fail to enter the membrane, whereas extremely hydrophobic molecules will enter readily but may have a hard time leaving it on the other side.
3.4.4 The membrane permeability of drugs can be improved by removing polar functional groups
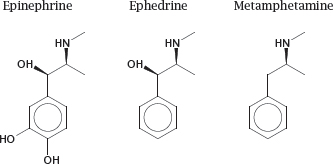
Notes: Removal of charged and polar functional groups can improve the ability of a drug molecule to cross lipid bilayers. For example, ephedrine and metamphetamine are analogues of epinephrine that have shed some hydroxyl groups. Accordingly, they penetrate cell membranes more readily than the parent compound, which promotes their uptake from the intestine and their distribution to the brain.
However, the structural changes also cause a shift in the molecular mode of action. Like epinephrine, both ephedrine and metamphetamine stimulate adrenergic synapses; however, while epinephrine acts directly on adrenergic receptors at the cell surface, ephedrine and metamphetamine act primarily on intracellular targets (see slide 6.14.3).
3.4.5 Resorption esters can improve the diffusion of drugs across membranes
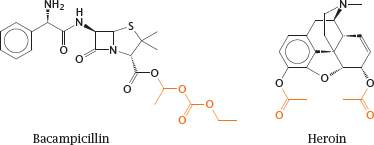
Notes: An alternate approach to improving the membrane penetration of drug molecules is to turn them into prodrugs, in which polar or ionizable groups are masked with hydrophobic residues. After uptake, these hydrophobic groups must be able to undergo enzymatic cleavage in order to release the original drug molecules. Most commonly, such functional groups are organic esters.
Bacampicillin is such a resorption ester; it contains ampicillin, which after uptake into the intestinal epithelium is released by esterases. Heroin is morphine with two acetyl groups added so as to mask two polar hydroxyl groups and facilitate distribution of the drug to the central nervous system. Once there, heroin undergoes cleavage by esterases, which releases the morphine again. In both prodrug molecules, the cleavable ester groups are highlighted.
3.4.6 History of heroin

Notes: Heroin is a semisynthetic drug, which is obtained from morphine through acetylation.1 It was originally intended not as a narcotic drug but rather as an analgesic milder than morphine that would be suitable for everyday use; and as these pictures illustrate, it was indeed marketed for a while in this vein. However, because of its improved penetration into the brain, it is actually stronger and more addictive than morphine.
3.4.7 Ionizable drug molecules may cross bilayers by non-ionic diffusion
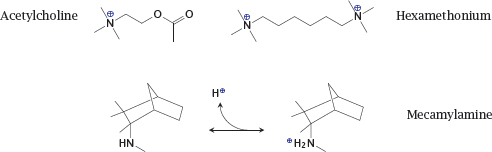
Notes: The physiological mediator acetylcholine possesses a quaternary amino group whose positive charge is essential for the interaction with the nicotinic acetylcholine receptor (NAR; see slide 6.10.1).
Hexamethonium is an inhibitor of the NAR. Since it contains two quaternary amino groups, it is transported across membranes very poorly and cannot be orally applied. In contrast, the inhibitor mecamylamine contains a tertiary amine. Therefore, it can cross bilayers in its unprotonated form and then bind a proton to acquire the charge necessary for receptor binding. This drug is amenable to uptake after oral ingestion. The transport across membranes of an ionizable drug in its non-ionized form is called non-ionic diffusion.
3.4.8 Gastric acid promotes accumulation of acetylsalicylic acid in the cells of the mucous membrane
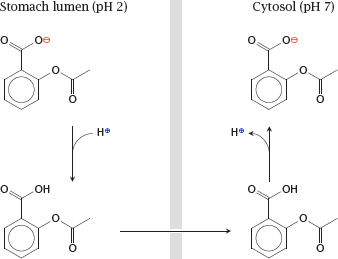
Notes: Non-ionic diffusion will be affected by pH gradients. The steepest such gradient exists at the surface of the gastric mucous membrane. The high proton concentration inside the stomach lumen can cause accumulation of acetylsalicylic acid inside the gastric epithelial cells and promote gastritis and ulcer formation.
To prevent the accumulation of acetylsalicylic acid in the gastric mucosal cells, the drug can be given an enteric coating, that is, be encapsulated in some sort of polymer that resists dissolution at the low pH in the stomach but readily dissolves in the slightly alkaline milieu of the small intestine (see slide 14.4.1).
3.5 Protein-mediated drug transport
While non-specific, passive diffusion is the most common mode of drug transport across cell membranes, many drugs are also subject to protein-mediated active or passive transport. An important class of membrane proteins involved in drug transport are the so called ABC transporters; ABC stands for ATP-binding cassette, which term refers to a structural motif that is conserved among these proteins. ABC transporters are found in many places, but prominently in the small intestine, the liver, the kidneys, and the blood brain barrier. Typically, they oppose the intracellular accumulation of drug molecules by extruding them back out of the cell, for example into the gut lumen or into the blood stream.
ABC transporters tend to have rather broad substrate specificities, and they can therefore transport/extrude a potentially large number of drugs. Overexpression of ABC transporter in tumor cells is an important mechanism of resistance to anticancer drugs. Similarly, ABC transporters in bacteria contribute to resistance to antibiotics.
Organic cation transporters and organic anion transporters are other major types of membrane proteins involved in drug transport, particularly in the kidneys and the liver. They are not powered by ATP, but some are powered by ion co-transport or antiport; that is, they perform secondary active transport. Like ABC transporters, organic anion and cation transporters tend to have broad substrate specificities.
3.5.1 Inward- and outward-facing conformations of ABC transporters