Chapter 7
Innate Immunity
Inflammation
Neal S. Rote, Sue E. Huether and Kathryn L. McCance

People are exposed daily to an environment containing a large variety of toxic substances and potentially infectious and disease-causing microorganisms. Without an efficient system of protection most individuals would succumb to these hazards early in life. That system consists of multiple complementary and interdependent layers. An outer layer of specialized epithelium, including the skin and mucosal surfaces, is relatively resistant to most environmental hazards and resists infection with disease-causing microorganisms.1 If the epithelial barrier is damaged, a highly efficient local and systemic response (inflammation) is mobilized to limit the extent of damage, protect against infection, and initiate repair of the damaged tissue. The natural epithelial barrier and inflammation confer innate resistance and protection, commonly referred to as innate, native, or natural immunity. Inflammation associated with infection usually initiates an adaptive process that results in a long-term and very effective immunity to the infecting microorganism, referred to as adaptive, acquired, or specific immunity. Adaptive immunity is relatively slow to develop but has memory and more rapidly targets and eradicates a second infection with a particular disease-causing microorganism.
The information presented in this chapter introduces the components and processes of innate immunity and sets the stage for Chapter 8, which discusses adaptive immunity. Although inflammation and adaptive immunity provide protection, either genetic or acquired aberrations in these processes can lead to disease. Diminution of innate or adaptive immunity may lead to critically decreased resistance to infection. Excessive inflammation or adaptive immunity may lead to damage to normal tissue or organs. Both may result in severe and potentially fatal disease, examples of which are discussed in Chapter 9. Many microorganisms that cause disease have developed methods of bypassing our protective systems. These are discussed in Chapter 10. Each chapter is designed to render an overview and is not intended to be all-inclusive. Protective mechanisms consist of a very large number of soluble factors and cells and would require many more pages to discuss in adequate detail. Different classes or groups of molecules and cells will be discussed, but only a few examples will be described in detail. Some components directly participate in the protective response, whereas others are designed to limit the extent of the response.
Human Defense Mechanisms
Innate immunity includes two lines of defense: natural barriers and inflammation (Table 7-1). Natural barriers are physical, mechanical, and biochemical barriers at the body’s surfaces and are in place at birth to prevent damage by substances in the environment and thwart infection by pathogenic microorganisms. If the surface barriers are breached, the second line of defense, the inflammatory response, is activated to protect the body from further injury, prevent infection of the injured tissue, and promote healing. The inflammatory response is a rapid activation of biochemical and cellular processes that is relatively nonspecific, with similar responses being initiated against a wide variety of causes of tissue damage.
TABLE 7-1
| CHARACTERISTICS | INNATE IMMUNITY | ADAPTIVE (ACQUIRED) IMMUNITY | |
| BARRIERS | INFLAMMATORY RESPONSE | ||
| Level of defense | First line of defense against infection and tissue injury | Second line of defense; occurs as a response to tissue injury or infection | Third line of defense; initiated when innate immune system signals the cells of adaptive immunity |
| Timing of defense | Constant | Immediate response | Delay between primary exposure to antigen and maximum response; immediate against secondary exposure to antigen |
| Specificity | Broadly specific | Broadly specific | Response is very specific toward “antigen” |
| Cells | Epithelial cells | Mast cells, granulocytes (neutrophils, eosinophils, basophils), monocytes/macrophages, natural killer (NK) cells, platelets, endothelial cells | T lymphocytes, B lymphocytes, macrophages, dendritic cells |
| Memory | No memory involved | No memory involved | Specific immunologic memory by T and B lymphocytes |
| Peptides | Defensins, cathelicidins, collectins, lactoferrin, bacterial toxins | Complement, clotting factors, kinins | Antibodies, complement |
| Protection | Protection includes anatomic barriers (i.e., skin and mucous membranes), cells and secretory molecules or cytokines (e.g., lysozymes, low pH of stomach and urine), and ciliary activity | Protection includes vascular responses, cellular components (e.g., mast cells, neutrophils, macrophages), secretory molecules or cytokines, and activation of plasma protein systems | Protection includes activated T and B lymphocytes, cytokines, and antibodies |
First Line of Defense: Physical, Mechanical, and Biochemical Barriers
Physical and Mechanical Barriers
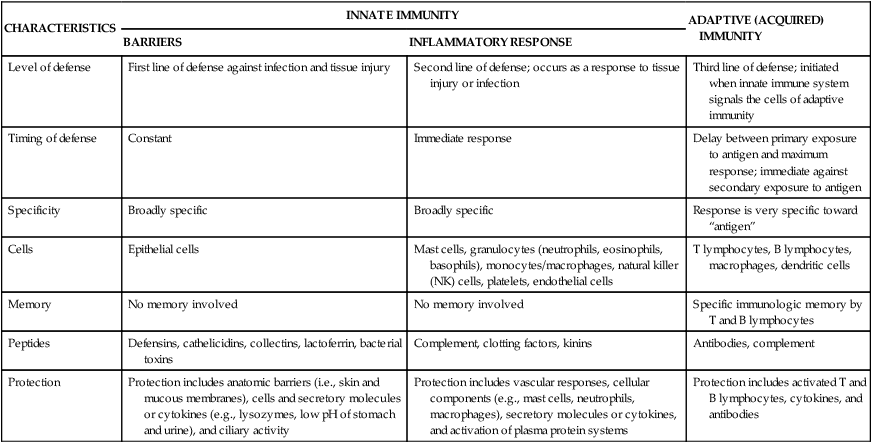
The physical barriers that protect against damage and infection are composed of tightly associated epithelial cells including those of the skin and of the membranous sheets lining the gastrointestinal, genitourinary, and respiratory tracts (Figure 7-1). The mucosal epithelial cells are highly interconnected junctions that prohibit the passage of microorganisms into the underlying tissue.2 The normal turnover of the cells in these sites as well as mechanisms for “washing” the surfaces may mechanically remove many infectious microorganisms and prevent their residence on the epithelial surfaces. For instance, the routine sloughing off and replacement of dead skin cells also removes adherent bacteria. Mechanical cleansing of the surfaces includes vomiting and urination. Goblet cells of the upper respiratory tract produce mucus that coats the epithelial surface and traps microorganisms that are removed by hairlike cilia that mechanically move the mucus upward to be expelled by coughing or sneezing. Additionally, the low temperature on the skin generally inhibits microorganisms, most of which prefer temperatures near 37° C for more efficient growth.
The digestive, respiratory, and genitourinary tracts and the skin form closed barriers between the internal organs and the environment. (From Grimes DE: Infectious diseases, St Louis, 1991, Mosby.)
Biochemical Barriers
Epithelial-Derived Chemicals
The epithelial surfaces of the body secrete a complex array of proteins that destroy potential pathogens. Epithelial cells secrete small-molecular-weight antimicrobial peptides.3 These are generally positively charged polypeptides of approximately 15 to 95 amino acids and can be divided into two classes—cathelicidins and defensins—based on their 3-dimensional structures. Both classes are in very high local concentrations and are toxic to several bacteria, fungi, and viruses. Cathelicidins have a linear α-helical shape, and only one is currently known to function in humans. In contrast, about 50 different defensins have been identified thus far. All are triple-stranded β-sheet structures. Defensin molecules contain 3 intrachain disulfide bonds and can be further subdivided into α (at least 6 identified in humans) and β types (at least 10 identified, but perhaps up to 40 different molecules), depending on how the cysteine residues are connected during formation of the disulfide linkages.4 The α-defensins often require activation by proteolytic enzymes, whereas the β-defensins are synthesized in active forms. Bacteria have cholesterol-free cell membranes, which may allow cathelicidins to insert into and disrupt their membranes. Given the similarity in their chemical charges, defensins may kill bacteria in the same way. These same chemicals also may contribute to other means of protection because they are also produced by monocytes, macrophages, and neutrophils, which are components of the inflammatory response. Cathelicidin is stored in neutrophils, mast cells, and a variety of epithelial cells. The α-defensins are particularly rich in the granules of neutrophils and may contribute to the killing of bacteria by those cells. They are also found in Paneth cells lining the small intestine, where they protect against a variety of disease-causing microorganisms. The β-defensins are found in a variety of epithelial cells lining the respiratory, urinary, and intestinal tracts, as well as in the skin. In addition to antibacterial properties, β-defensins may also help protect epithelial surfaces from human immunodeficiency virus (HIV) infection. Both classes of antimicrobial peptides also can activate cells of innate and adaptive immunity.
Bacteria-Derived Chemicals
The body’s surfaces are colonized with a spectrum of microorganisms, the normal microbiome. Each surface, including the skin and the mucous membranes of the eyes, upper and lower gastrointestinal tracts, urethra, and vagina, is colonized by a combination of mostly bacteria and occasionally fungi that is unique to the particular location. The microorganisms in the microbiome do not normally cause disease, and although their relationship with humans has been referred to as commensal (to the benefit of one organism without affecting the other), the relationship may be more mutualistic (to the benefit of both organisms). Using the colon for an example, at birth the lower gut is relatively sterile but colonization with bacteria begins quickly, with the number, diversity, and concentration increasing progressively during the first year of life. To the benefit of humans, many of these microorganisms help digest fatty acids, large polysaccharides, and other dietary substances; produce biotin and vitamin K; and assist in the absorption of various ions, such as calcium, iron, and magnesium.5
These bacteria contribute to our innate protection against pathogenic microorganisms in the colon. They compete with pathogens for nutrients and block attachment to the epithelium. Members of the normal microbiome also produce chemicals (ammonia, phenols, indoles, and other toxic materials) and toxic proteins (bacteriocins) that inhibit colonization by pathogenic microorganisms. Prolonged treatment with broad-spectrum antibiotics can alter the normal intestinal microbiome, decreasing its protective activity, and lead to an overgrowth of pathogenic microorganisms, such as the yeast Candida albicans or the bacteria Clostridium difficile (overgrowth can cause pseudomembranous colitis, an infection of the colon). Additionally, the normal microbiome of the gut help train the adaptive immune system by inducing the growth of gut-associated lymphoid tissue (where cells of the adaptive immune system reside) and the development of both local and systemic adaptive immune systems.6
The bacterium Lactobacillus is a major constituent of the normal vaginal microbiome in healthy women.7 This microorganism produces a variety of chemicals (e.g., hydrogen peroxide, lactic acid, bacteriocins) that help prevent infections of the vagina and urinary tract by other bacteria and yeast. Prolonged antibiotic treatment can diminish colonization with Lactobacillus and increase the risk for urologic or vaginal infections, such as vaginosis.
if the individual’s defenses are compromised. These microorganisms are normally controlled by the innate and adaptive immune systems and contribute to our defenses. For example, Pseudomonas aeruginosa is a member of the normal microbiome of the skin and produces a toxin that protects against infections with staphylococcal and other bacteria. However, severe burns compromise the integrity of the skin and may lead to life-threatening systemic pseudomonal infections.
Second Line of Defense: The Inflammatory Response
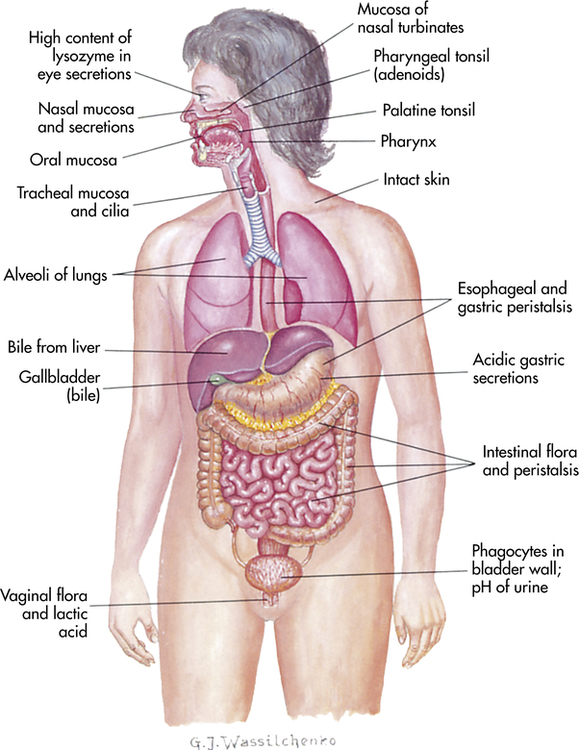
If cells and tissues are damaged the inflammatory response is usually activated (Figure 7-2). Injury can have a variety of causes including infection, mechanical damage, oxygen deprivation (ischemia), nutrient deprivation, genetic or immune defects, chemical agents, temperature extremes, or ionizing radiation. Inflammation (1) depends on the activity of both cellular and chemical components, and (2) is nonspecific, meaning that it takes place in approximately the same way regardless of the type of stimulus or whether exposure to the same stimulus has occurred in the past.
Inflammation is usually initiated by cellular injury, which results in mast cell degranulation, the activation of three plasma systems, and the release of subcellular components from the damaged cells. These systems are interdependent, so that induction of one (e.g., mast cell degranulation) can result in activation of the other two. The result is the development of microscopic changes in the inflamed site, as well as characteristic clinical manifestations. The figure numbers refer to those in which more detailed information may be found on that portion of the response.
Vascular Response
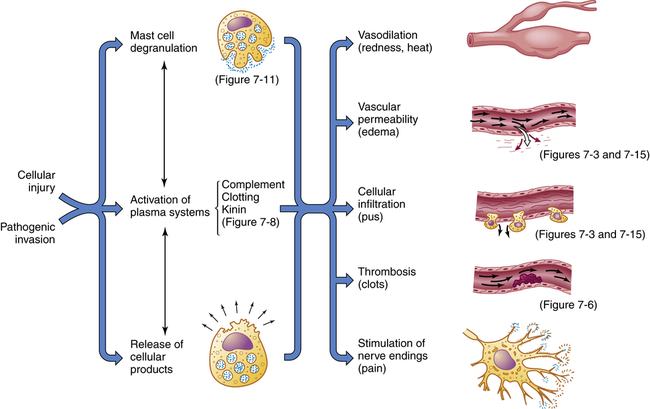
Inflammation occurs in tissue that has a blood supply (vascularized) and results in a group of easily observable characteristics: redness, heat, swelling, and pain. This tetrad represents the “cardinal signs of inflammation” and was identified in the first century by a Roman writer, Celsus. Microscopically, inflammatory changes occur at the vascular level (Figure 7-3). The three characteristic changes in the microcirculation (arterioles, capillaries, and venules) near the site of an injury include the following:
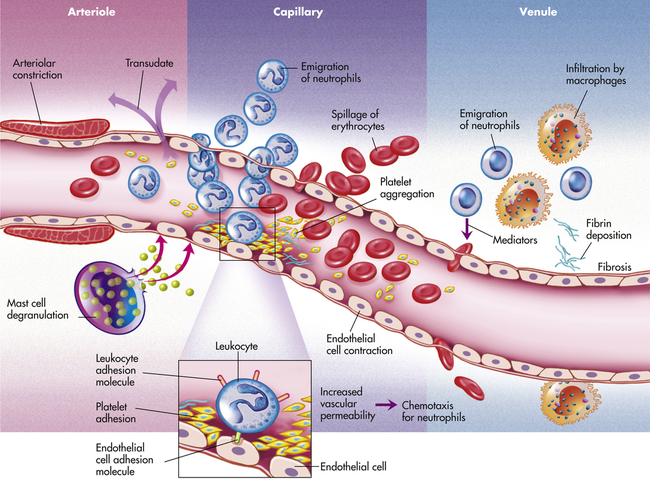
1. Blood vessel dilation (vasodilation)
2. Increased vascular permeability and leakage of fluid out of the vessel
3. White blood cell adherence to the inner walls of vessels and their migration through vessel walls to the site of injury (diapedesis)
The effects of inflammation are visible within seconds. First, arterioles near the site of infection or injury constrict briefly. Vasodilation then causes slower blood velocity and increases local blood flow to the injured site. The increased flow and capillary permeability result in leakage of plasma from the vessels, causing swelling (edema) in the surrounding tissue. As plasma moves outward, blood remaining in the microcirculation flows more slowly and becomes more viscous. The increased blood flow and increasing concentration of red cells at the site of inflammation cause locally increased warmth and redness. Leukocytes adhere to vessel walls. At the same time, biochemical mediators (e.g., histamine, bradykinins, leukotrienes, prostaglandins) stimulate the endothelial cells that line capillaries and venules to retract, creating spaces at junctions between the cells, allowing leukocytes and plasma to enter the surrounding tissue (intercellular junctions are described in Chapter 1).
1. Prevent infection and further damage by contaminating microorganisms through the influx of fluid to dilute toxins produced by bacteria and released from dying cells, the influx and activation of plasma protein systems that help destroy and contain bacteria (e.g., complement system, clotting system), and the influx of cells (e.g., neutrophils, macrophages) that “eat” and destroy cellular debris and infectious agents.
2. Limit and control the inflammatory process through the influx of plasma protein systems (e.g., clotting system), plasma enzymes, and cells (e.g., eosinophils) that prevent the inflammatory response from spreading to areas of healthy tissue.
3. Interact with components of the adaptive immune system to elicit a more specific response to contaminating pathogen(s) through the influx of macrophages and lymphocytes.8
4. Prepare the area of injury for healing through removal of bacterial products, dead cells, and other products of inflammation (e.g., by way of channels through the epithelium or drainage by lymphatic vessels) and initiation of mechanisms of healing and repair.
Fluid and debris that accumulate at an inflamed site are drained by lymphatic vessels. This process also facilitates the development of adaptive immunity because microbial antigens in lymphatic fluid pass through the lymph nodes, where they activate both B and T lymphocytes. (This process is discussed in Chapter 8, and the lymphatic system is described in Chapter 27.)
Inflammation and repair can be divided into several phases (Figure 7-4). The characteristics of the early (i.e., acute) inflammatory response differ from those of the later (i.e., chronic) response, and each phase involves different biochemical mediators and cells that function together. The acute inflammatory response is of short duration; that is, it continues only until the immediate threat to the host is eliminated. This usually takes 8 to 10 days from onset to healing. The acute inflammatory response begins immediately after cellular injury or infection occurs and involves a vascular response, activation of plasma protein systems, and activation of a variety of cells. (Mechanisms of cellular injury are described in Chapter 2.)
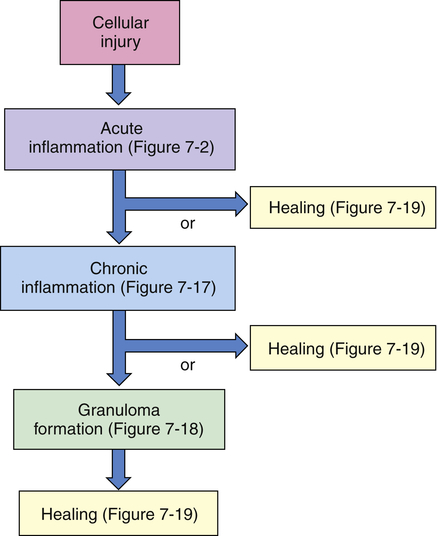
Cellular injury leads to acute inflammation and may result in resolution and healing of the injured site or may progress into chronic inflammation. Chronic inflammation in turn may result in healing or progress to development of a granuloma. The final step of the inflammatory process is usually healing and reconstruction of the damaged tissue. The figure numbers refer to those in which more detailed information on that portion of the process may be found.
Plasma Protein Systems
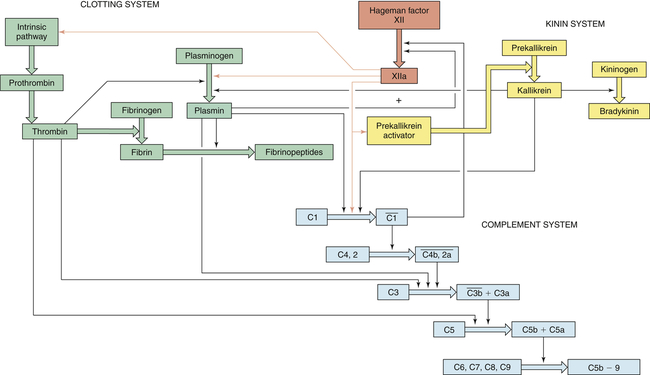
Three key plasma protein systems are essential to an effective inflammatory response. These are the complement system, the clotting system, and the kinin system (Figures 7-5, 7-6, and 7-7, respectively). Although each system has a unique role in inflammation, they also have many similarities. Each system consists of multiple proteins in the blood. To prevent activation in unnecessary situations, each protein is normally in an inactive form. Several of the proteins are enzymes that circulate in inactive forms as proenzymes. Each system contains a few proteins that can be activated by products of tissue damage or infection. Activation of the first component of a system results in sequential activation of other components, leading to a biologic function that helps protect the individual. This sequential activation is referred to as a cascade. Thus, we refer to the complement cascade, the clotting cascade, or the kinin cascade. In some cases, activation of a protein may require that it be enzymatically cut into two pieces or fragments of different size. Usually the larger fragment continues the cascade by activating the next component, and the smaller fragment frequently has potent biologic activities to promote inflammation.
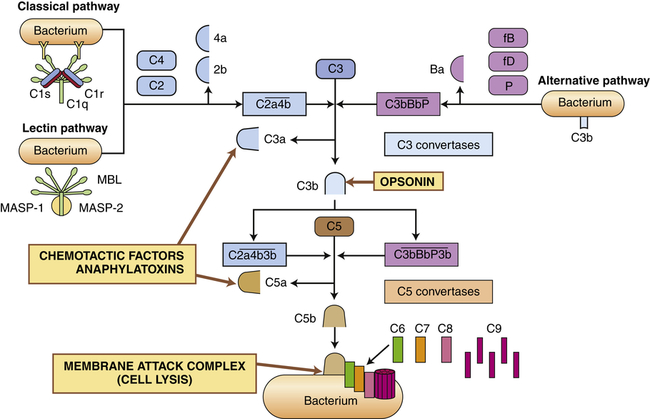
The complement system is activated by three pathways: the classical pathway, the lectin pathway, and the alternative pathway. During activation, many complement components are cleaved into fragments (2b, 4a, Ba, C3a, and C5a). The smaller fragments frequently have potent biologic activities and may serve as chemotactic factors and anaphylatoxins. The larger activated fragments are usually converted into active enzymes (indicated by the bar above the names) and form complexes with additional components in the cascade. The classical pathway is usually activated by antigen-antibody complexes through component C1, which consists of C1q and two C1r and C1s molecules. As indicated, the C1q must simultaneously bind to two antibody molecules (indicated by Y-shaped structures). The lectin pathway is activated by mannose-binding lectin (MBL), which binds to two mannose-rich pathogen-associated molecular patterns on the surface of a bacterium. MBL contains two associated enzymes, MASP-1 and MASP-2, and functions in a manner similar to C1. C1 and MBL each activate complement components C4 and C2. The alternative pathway is activated by many agents, such as bacterial polysaccharides, which bind and stabilize C3b, which is produced by normal breakdown of C3 in the blood. The C3b forms the site of binding of factor B (fB), which is activated by factor D (fD) into Bb and the small fragment Ba. Properdin (P) helps stabilize the complex. Each pathway produces C3 and C5 convertases, which are enzymatically active complexes that activate C3 and C5, respectively. C3b produced by the C3 convertase can function as an opsonin. C5b initiates assemblage of the membrane attack complex (MAC), which results in multiple C9 molecules forming a pore in the bacterial membrane.
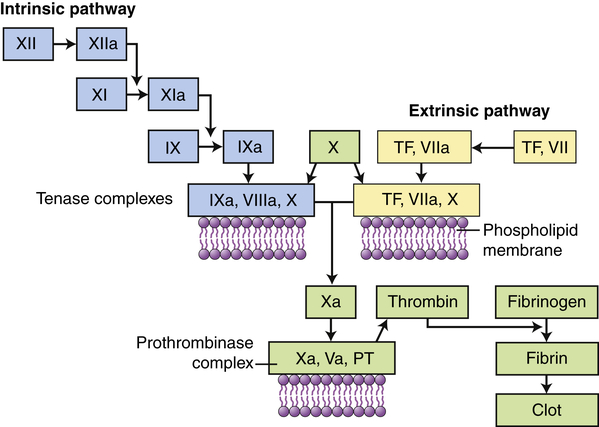
Clotting is activated through two pathways: the intrinsic pathway and the extrinsic pathway. The intrinsic pathway is initiated by the activation of Hageman factor (XII) into XIIa (activated factors are enzymes and are indicated by a lowercase a). The sequential activation of other intrinsic pathway components results in formation of a complex of IXa, VIIIa, and X. The extrinsic pathway is activated by exposure of tissue factor (TF) during tissue damage. TF complexes with factor VII, which is activated (VIIa) and forms a complex with factor X (TF, VIIa, X). Both the intrinsic and the extrinsic pathway complexes are dependent on calcium, form on phospholipid membranes that are rich in phosphatidylserine, and have “tenase” activity (can activate factor X into Xa). Factor X begins a common pathway in which Xa complexes with Va and prothrombin (PT), with calcium and phospholipid membranes, to form an active prothrombinase (activates prothrombin into thrombin). Thrombin is an enzyme that cuts high-molecular-weight fibrinogen into fibrin molecules. Fibrin polymerizes to form a clot.
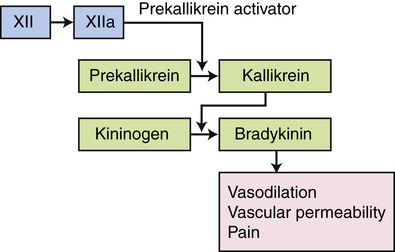
The kinin pathway is activated by factor XIIa from the clotting system, which functions as an enzyme (prekallikrein activator) to convert prekallikrein into kallikrein. Enzymatically active kallikrein converts kininogen into bradykinin.
Complement System
1. Classical pathway: activated by proteins of the adaptive immune system (antibodies) bound to their specific targets (antigen)
2. Lectin pathway: activated by mannose-containing bacterial carbohydrates
3. Alternative pathway: activated by gram-negative bacterial and fungal cell wall polysaccharides
The principal routes by which the complement cascade may be activated are shown in Figure 7-5.
Activation of the classical pathway begins with the activation of complement protein C1 and is preceded by formation of a complex between an antigen and an antibody to form an antigen-antibody complex (immune complex) (discussed in Chapter 8). The antigen may be a unique chemical component of the surface of a bacterium or other microorganism. Most pathogens express multiple antigens; therefore, multiple antibodies are usually bound in the complex. The first component of the classical complement cascade, C1, has six sites that can bind to antibodies, and efficient activation of the complement cascade usually requires concurrent binding of C1 to at least two antibody molecules. The complex formed by antigen-antibody-complement binding is shown in Figure 7-5. C1 is a macromolecular complex consisting of C1q and two molecules each of C1r and C1s. A conformational change in C1 results in an enzymatically active molecule whose substrates are C4 and C2. The resultant complex formed by the interaction of C1, C4, and C2 uses C3 as a substrate, resulting in the production of C3a and C3b. A complex that has C3 as a substrate is generally referred to as a C3 convertase. The addition of C3b to the complex changes the substrate specificity to C5, resulting in the conversion of C5 to C5a and C5b. A complex that has C5 as a substrate is generally called a C5 convertase. Thus activation of C1 initiates the sequential enzymatic activation of all other components of the classical pathway, ultimately resulting in the activation of C5. The classical pathway also can be activated to a lesser degree by biologic molecules other than antibody, including heparin (a charged molecule that prevents clotting), deoxyribonucleic acid (DNA) or ribonucleic acid (RNA), and C-reactive protein, which is increased in the blood during inflammation.
The most important result of complement activation is the production of fragments during the activation of C4, C2, C3, and C5. The fragments C4a, C2b, C3a, and C5a are soluble and of low-molecular-weight that contribute in other ways to the inflammatory response. C2b affects smooth muscle, causing vasodilation and increased vascular permeability. C3a and C5a, and to a limited extent C4a, are anaphylatoxins; that is, they induce rapid mast cell degranulation (release of granular contents) and the release of histamine (see Figure 7-11, p. 206), causing vasodilation and increased capillary permeability.9 C5a is the major chemotactic factor for neutrophils. C3a is approximately 100 times less potent in chemotactic and anaphylatoxic activity. A chemotactic factor is a biochemical substance that attracts leukocytes to the site of inflammation.
Thick colored arrows denote the activation of factors within a system. Thin arrows denote where a particular factor activates another system.
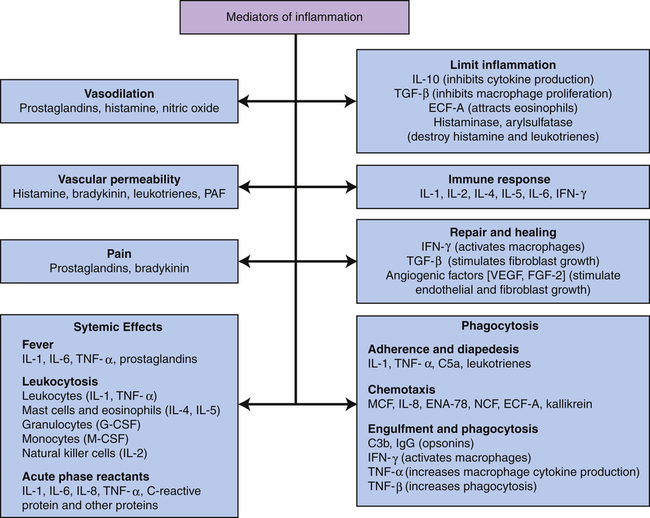
C3b, Large fragment produced from complement component C3; C5a, small fragment produced from complement component C5; ECF-A, eosinophil chemotactic factor of anaphylaxis; ENA, epithelial-dermoid neutrophil attractant; FGF, fibroblast growth factor; G-CSF, granulocyte colony-stimulating factor; IFN, interferon; IgG, immunoglobulin G (predominant class of antibody in the blood); IL, interleukin; MCF, monocyte chemotactic factor; M-CSF, monocyte colony-stimulating factor; NCF, neutrophil chemotactic factor; PAF, platelet-activating factor; TGF, T-cell growth factor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
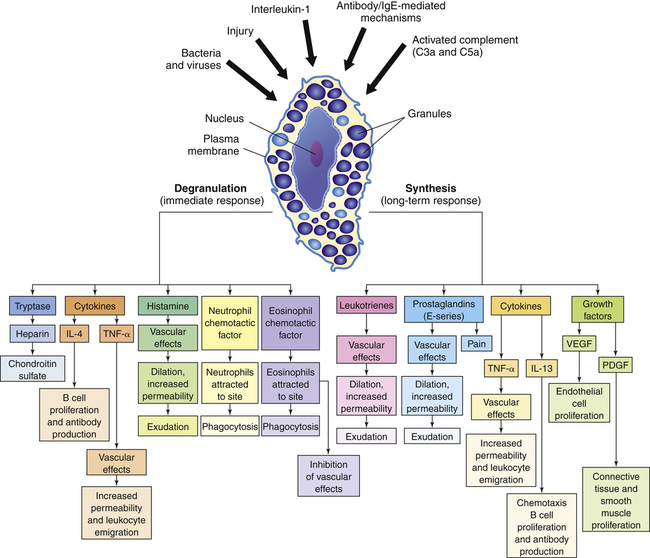
The depiction of a tissue mast cell shows darkly stained granules in the cytoplasm. IL-4, Interleukin-4; IL-13, interleukin-13; PDGF, platelet-derived growth factor; TNF-α, tumor necrosis factor-alpha; VEGF, vascular endothelial growth factor.
C3b adheres to the surface of a pathogenic microorganism and serves as an efficient opsonin. Opsonins are molecules that “tag” microorganisms for destruction by cells of the inflammatory system (primarily neutrophils and macrophages [see pp. 208 and 209]). C3b on the cell surface also can be broken down by several enzymes in the blood into inactive fragments (e.g., iC3b), which retain opsonic activity.
Clotting System
The clotting system can be activated by many substances that are released during tissue injury and infection, including collagen, proteinases, kallikrein, and plasmin, as well as by bacterial products such as endotoxins. Like the complement cascade, the coagulation cascade can be activated through different convergent pathways (see Figure 7-6). The tissue factor (extrinsic) pathway is activated by tissue factor (TF) (also called tissue thromboplastin) that is released by damaged endothelial cells in blood vessels and reacts with activated factor VII (VIIa). The contact activation (intrinsic) pathway is activated when the vessel wall is damaged and Hageman factor (factor XII) in plasma contacts negatively charged subendothelial substances. The pathways converge at factor X. Activation of factor X begins a common pathway leading to activation of fibrin that polymerizes to form a fibrin clot. The coagulation system is discussed in more detail and illustrated again in Chapter 27.
Kinin System
The third plasma protein system, the kinin system, augments inflammation in several ways. The primary kinin produced from the kinin system is bradykinin, which causes dilation of blood vessels, acts with prostaglandins to stimulate nerve endings and induce pain, causes smooth muscle cell contraction, increases vascular permeability, and may increase leukocyte chemotaxis (see Figure 7-2). Bradykinin induces smooth muscle contraction more slowly than histamine and, along with prostaglandins of the E series, is probably responsible for endothelial cell retraction and increased vascular permeability in the later phases of inflammation (endothelial cell retraction is shown in Figures 7-3 and 7-15).
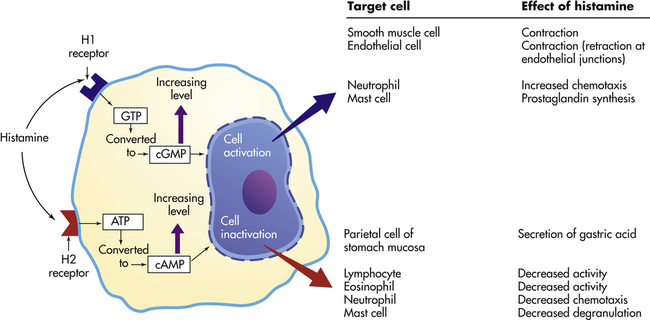
Effects depend on (1) density and affinity of H1 or H2 receptors on the target cell, and (2) the identity of the target cell. ATP, Adenosine triphosphate; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; GTP, guanosine triphosphate.
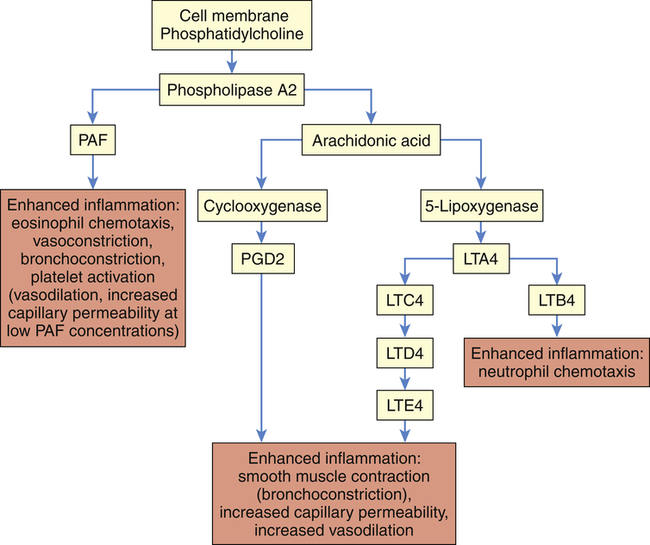
LTA4, LTC4, LTD4, LTE4, LTB4, Various leukotriene molecules; PAF, platelet-activating factor; PGD2, prostaglandin D2.
The lymphocytes are small and spherical; the macrophages are larger and more irregular in shape. (From Raven PH, Johnson GB: Biology, St Louis, 1992, Mosby.)
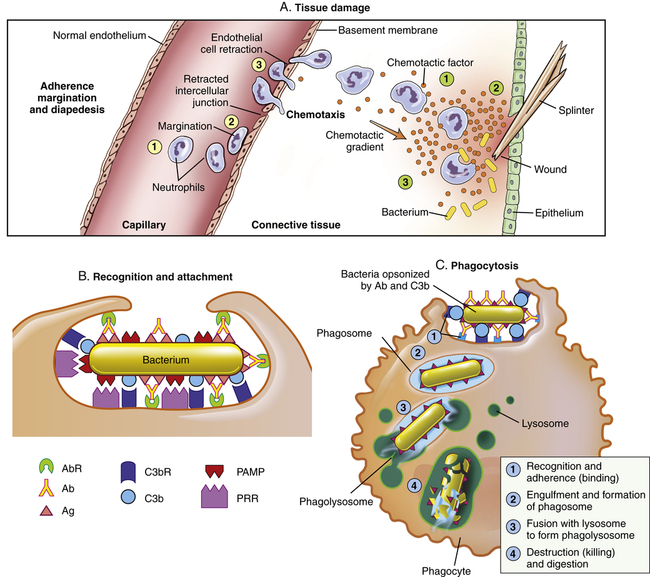
The process that results in phagocytosis is characterized by three interrelated steps: adherence and diapedesis, tissue invasion by chemotaxis, and phagocytosis. A, Tissue damage. Adherence, margination, and diapedesis: The primary phagocyte in the blood is the neutrophil, which usually moves freely within the vessel (1). At sites of inflammation, the neutrophil progressively develops increased adherence to the endothelium, leading to accumulation along the vessel wall (margination or pavementing) (2). At sites of endothelial cell retraction the neutrophil exits the blood by means of diapedesis (3). Chemotaxis: In the tissues, the neutrophil detects chemotactic factor gradients through surface receptors (1) and migrates towards higher concentrations of the factors (2). The high concentration of chemotactic factors at the site of inflammation immobilizes the neutrophil (3). B, Recognition and attachment. Specific receptors and ligands for recognition and attachment. C, Phagocytosis. (1) Opsonized microorganisms bind to the surface of a phagocyte through specific receptors. (2) The microorganism is engulfed (ingested) into a phagocytic vacuole, or phagosome. (3) Lysosomes fuse with the phagosome, resulting in the formation of a phagolysosome. During this process the microorganism is exposed to products of the lysosomes, including a variety of enzymes and products of the hexose-monophosphate shunt (e.g., H2O2,
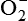 ). (4) The microorganism is killed and digested. Ab, Antibody; AbR, antibody receptor; Ag, antigen; C3b, complement component C3b; C3bR, complement C3b receptor; PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor.
). (4) The microorganism is killed and digested. Ab, Antibody; AbR, antibody receptor; Ag, antigen; C3b, complement component C3b; C3bR, complement C3b receptor; PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor.The kinin system is activated by stimulation of the plasma kinin cascade (see Figure 7-7). The conversion of plasma prekallikrein to kallikrein is induced by prekallikrein activator, which is identical to factor XIIa (the product that results from activation of Hageman factor—factor XII) of the clotting cascade. Kallikrein then converts kininogen to bradykinin. Although the plasma kinin cascade is one pathway that leads to the production of bradykinin, tissue kallikreins in saliva, sweat, tears, urine, and feces provide other sources for this inflammatory mediator. These tissue kallikreins convert serum kininogens to kallidin, also known as Lys-bradykinin, which may be converted to bradykinin by plasma aminopeptidase. In order to limit the extent of inflammation, kinins are rapidly degraded by kininases, enzymes present in plasma and tissues.
Interactions Among the Plasma Protein Systems
The three plasma protein systems are highly interactive so that activation of one system results in the production of a large number of very potent, biologically active substances that further activate the other systems (Figure 7-8). Very tight control of these processes is essential for two reasons:
1. The inflammatory process is critical for an individual’s survival; thus, efficient activation must be guaranteed regardless of the cause of tissue injury.
2. The biochemical mediators generated during these processes are so potent and potentially detrimental to the host itself that their actions must be strictly confined to injured or infected tissues only.
1. Activation of the clotting cascade through factor XI
2. Control of clotting through conversion of plasminogen proactivator to plasminogen activator, resulting in the generation of plasmin
3. Activation of the kinin system by activated Hageman factor (prekallikrein activator)
Many enzymes from the plasma regulate the activity of these pathways, such as carboxypeptidase that inactivates the anaphylatoxic activities of C3a and C5a, and kininases that degrade kinins. Many other natural inhibitors are present, including enzymes that degrade histamine (histaminase), activated complement components, kallikrein, and plasmin. Another example of a common regulator is C1-esterase inhibitor (C1-inh).10 C1-inh inhibits complement activation through reactivity with C1 (classical pathway), MASP-2 (lectin pathway), and C3b (alternative pathway). It is also a major inhibitor of the clotting and kinin pathways (e.g., kallikrein, activated Hageman factor XIIa). A genetic defect in C1-inh (C1-inh deficiency) results in hereditary angioedema, which is a self-limiting edema of cutaneous and mucosal layers resulting from stress, illness, or relative minor or unapparent trauma. The disease is characterized by hyperactivation of all three plasma protein systems, although excessive production of bradykinin appears to be the principal cause of increased vascular permeability.
Cellular Mediators of Inflammation
Inflammation is a process in vascular tissue; thus the cellular components of inflammation are found in the blood and in the tissue surrounding the blood vessels. The vessels are lined with endothelial cells, which under normal conditions actively maintain normal blood flow. During inflammation the vascular endothelium becomes a principal coordinator of blood clotting and the passage of cells and fluid into the tissue. The tissue close to the vessels contains mast cells, which are probably the most important activators of inflammation. The tissue also contains dendritic cells, which connect the innate and adaptive immune responses. The blood contains a complex mixture of cells (see Figure 7-3). Blood cells are divided into erythrocytes (red blood cells), platelets, and leukocytes (white blood cells). Erythrocytes carry oxygen to the tissues, and platelets are small cell fragments involved in blood clotting. Leukocytes are subdivided into granulocytes (containing many enzyme-filled cytoplasmic granules), monocytes, and lymphocytes. Granulocytes are the most common leukocytes and are classified by the type of stains needed to visualize their granules (basophils, eosinophils, and neutrophils). Monocytes are precursors of macrophages that are found in the tissues. Various forms of lymphocytes participate in the innate (natural killer [NK] cells) and the adaptive immune response (B and T cells).
Cellular Receptors
Cells of both innate and adaptive immunity must recognize and respond to their environment, whether to products of damaged cells or to potential pathogenic microorganisms. Each cell has receptors on the cell surface that specifically bind soluble substances (ligands) produced during tissue damage or infection. The binding of a ligand to its receptor results in activation of intracellular signaling pathways and activation of the cell. As will be discussed in Chapter 8, B and T lymphocytes of the adaptive immune system have evolved surface receptors (i.e., the T-cell receptor, or TCR, and the B-cell receptor, or BCR) that bind a large spectrum of antigens. Cells involved in innate resistance have evolved a different set of receptors that recognize a much more limited array of specific molecules. These are referred to as pattern recognition receptors (PRRs), and they recognize molecular “patterns” on infectious agents or their products (pathogen-associated molecular patterns, or PAMPs), or products of cellular damage (necrosis or apoptosis; damage-associated molecular patterns, or DAMPs). PRRs are generally found on cells at the interface of the host and environment (i.e., skin, respiratory tract, gastrointestinal tract, genitourinary tract), where they monitor for products of cellular damage and potentially infectious microorganisms. Although most PRRs are on the cell surface, some are secreted or intracellular.11 An example of a secreted PRR is mannose-binding lectin of the lectin pathway of complement activation. Cellular PRRs include Toll-like receptors, complement receptors (CRs), scavenger receptors, glucan receptors, and mannose receptors. A group of intracellular protein complexes, called inflammasomes, function as PRRs within cells.12 Inflammasomes primarily bind cellular stress–related molecules, a type of DAMPs, and control the production of interleukin-1β (IL-1β).13
In humans, at least 11 different Toll-like receptors (TLRs) have been described, 10 of which are functional.14 They are expressed on the surface of many cells that have direct and early contact with potential pathogenic microorganisms. These include mucosal epithelial cells, mast cells, neutrophils, macrophages, dendritic cells, and some subpopulations of lymphocytes. (Dendritic cells are found in the skin, mucosa, and lymphoid tissues, where they have developed from Langerhans cells and function as highly specialized initiators of the adaptive immune response.) TLRs recognize a large variety of PAMPs located on the microorganism’s cell wall or surface (e.g., bacterial lipopolysaccharide [LPS], peptidoglycans, and lipoproteins, yeast zymosan, viral coat proteins), other surface structures (e.g., bacterial flagellin), or microbial nucleic acid (e.g., bacterial DNA, viral double-stranded RNA). Some TLRs recognize host factors that are produced by “stressed” or damaged cells (e.g., breakdown products of extracellular matrix proteins, chromatin). Interactions between PAMPs and TLRs, with the collaboration of other cellular receptors (e.g., CD14), can result in activation of the cell and the release of soluble products (e.g., cytokines) that increase local resistance to the pathogenic microorganism. TLRs are also one of the bridges between innate resistance and the adaptive immune response through the induction of cytokines that increase the response of lymphocytes to foreign antigens on the pathogens. Genetic polymorphisms in TLRs may explain some observed differences among individuals’ resistance and susceptibility to infections. Information on each of the TLRs found in humans is shown in Table 7-2.
TABLE 7-2
CELLULAR SOURCE AND MICROBIAL TARGET FOR EACH TOLL-LIKE RECEPTOR (TLR)
| RECEPTOR | CELLULAR EXPRESSION PATTERN | PAMP RECOGNITION |
| TLR1 | Cell surface (ubiquitous): neutrophils, monocytes/macrophages, dendritic cells, T cells, B cells, NK cells | Fungal, bacterial, viral; forms heterodimer with TLR2 (see TLR2 recognition) |
| TLR2 | Cell surface: neutrophils, monocytes/macrophages, dendritic cells | Fungal (yeast zymosan), bacterial (gram-positive bacterial peptidoglycan, lipoproteins), viral (lipoproteins) |
| TLR3 | Intracellular: monocytes/macrophages, dendritic cells, T cells, NK cells, epithelial cells | Double-stranded RNA produced by many viruses |
| TLR4 | Cell surface: granulocytes, monocytes/macrophages, dendritic cells, T cells, B cells, epithelial cells | Bacterial (primarily gram-negative bacterial LPS, lipoteichoic acids), viral (RSV F protein, hepatitis C) |
| TLR5 | Cell surface: granulocytes, monocytes/macrophages, dendritic cells, NK cells, epithelial cells | Bacterial (flagellin); forms heterodimer with TLR4 |
| TLR6 | Cell surface: monocytes/macrophages, dendritic cells, B cells, NK cells | Fungal, bacterial, viral; forms heterodimer with TLR2 (see TLR2 recognition) |
| TLR7 | Intracellular: monocytes/macrophages, dendritic cells, B cells | Natural ligand uncertain; may bind viral single-strand RNA |
| TLR8 | Cell surface: monocytes/macrophages, dendritic cells, NK cells | Natural ligand uncertain; may bind fungal PAMPs or viral single-stranded RNA |
| TLR9 | Intracellular: monocytes/macrophages, dendritic cells, B cells | Bacterial (unmethylated DNA [CpG dinucleotides]) |
| TLR10 | Cell surface: monocytes/macrophages, dendritic cells, B cells | Natural ligand uncertain; may form heterodimers with TLR2 |
| TLR11 | TLR11 gene does not code a full length protein in humans | No known immune response |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


