Figure 39–1 Bacterial protein synthesis and sites of drug action. The bacterial ribosome is composed of a 30S subunit and a 50S subunit. The steps in protein synthesis and translation of messenger RNA (mRNA) include the binding of aminoacyl transfer RNA (tRNA) to the ribosome, the formation of a peptide bond, and translocation. (A) Under normal circumstances, the nascent peptide is attached to the ribosome at the peptidyl site (P site), and the next aminoacyl tRNA binds to the acceptor or aminoacyl site (A site). Tetracyclines block aminoacyl tRNA from binding to the A site. Aminoglycosides and spectinomycin cause misreading of the genetic code, which leads to binding of the wrong aminoacyl tRNA and insertion of the wrong amino acid into the nascent peptide. (B) Macrolides, chloramphenicol, and dalfopristin block peptidyl transferase, the enzyme that catalyzes the formation of a peptide bond between the nascent peptide and the amino acid attached to the A site. (C) Macrolides and clindamycin block the translocation step in which the nascent peptide is transferred from the A site to the P site following the formation of a new peptide bond.
SITES OF DRUG ACTION
As shown in Figure 39–1, each type of antibiotic discussed in this chapter acts at a specific site on the ribosome to inhibit one or more steps in protein synthesis. Tetracyclines, aminoglycosides, and spectinomycin act at the 30S ribosomal subunit. Macrolides, chloramphenicol, dalfopristin, and clindamycin act at the 50S ribosomal subunit.
Tetracyclines competitively block binding of tRNA to the 30S subunit and thereby prevent the addition of new amino acids to the growing peptide chain. This reversibility of this effect accounts for the bacteriostatic action of tetracyclines.
Aminoglycosides and spectinomycin also bind to the 30S subunit, where they interfere with the initiation of protein synthesis and cause misreading of the genetic code so that the wrong amino acid is inserted into the protein structure. These irreversible actions account for the bactericidal effects of these antibiotics.
Macrolides, chloramphenicol, and dalfopristin block peptidyl transferase, the enzyme that catalyzes the formation of a peptide bond between the new amino acid and the nascent peptide. Macrolides and clindamycin prevent translocation of the nascent peptide from the acceptor or aminoacyl site (A site) to the peptidyl site (P site) on the ribosome which, in turn, prevents binding of the next aminoacyl tRNA to the ribosome.
DRUGS THAT AFFECT THE 30S RIBOSOMAL SUBUNIT
Aminoglycosides
The aminoglycosides include amikacin, gentamicin, neomycin, streptomycin, and tobramycin. The properties and major clinical uses of these drugs are compared in Table 39–1 and 39–2.
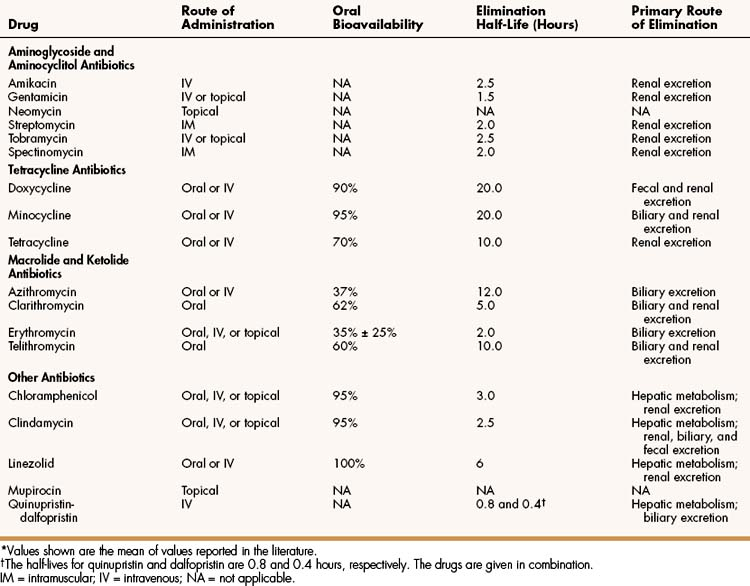
TABLE 39–2 Major Clinical Uses of Selected Bacterial Protein Synthesis Inhibitors
| Drug | Infections |
|---|---|
| Aminoglycosides | |
| Streptomycin | Plague, tularemia, drug-resistant tuberculosis |
| Gentamicin, tobramycin, and amikacin | Serious infections due to aerobic gram-negative bacilli, including Pseudomonas aeruginosa and many Enterobacteriaceae species |
| Tetracycline antibiotics | Lyme disease, Rocky Mountain spotted fever, ehrlichiosis, granuloma inguinale, brucellosis, cholera, relapsing fever, peptic ulcer disease due to H. pylori, chlamydial urethritis, acne, MRSA |
| Erythromycin | Respiratory tract infections due to streptococci, pneumococci, Legionella pneumophila, Mycoplasma pneumoniae, or Chlamydia pneumoniae |
| Azithromycin, clarithromycin | Respiratory tract infections due to organisms sensitive to erythromycin, Haemophilus influenzae, or Moraxella catarrhalis, Mycobacterium avium-intracellulare |
| Clarithromycin | Peptic ulcer disease due to Helicobacter pylori |
| Telithromycin | Community acquired pneumonia due to pneumococci, Legionella, Chlamydia, and other organisms |
| Chloramphenicol | Meningitis, brain abscess |
| Clindamycin | Streptococcal, staphylococcal, and anaerobic infections |
| Quinupristin-dalfopristin | Infections due to vancomycin-resistant Enterococcus faecium |
| Linezolid | Infections due to vancomycin-resistant Enterococcus faecium, streptococci, and methicillin-resistant staphylococci |
| Mupirocin | Impetigo due to streptococci, staphylococciEradication of nasal colonization of MRSA |
MRSA = methicillin-resistant Staphylococcus aureus.
CHEMISTRY AND PHARMACOKINETICS
Aminoglycoside antibiotics are composed of amino sugars linked through glycosidic bonds. The amino groups are highly basic and become extensively protonated and ionized in body fluids. For this reason, the aminoglycosides are poorly absorbed from the gut and must be administered parenterally for the treatment of systemic infections. Occasionally, they are administered orally to treat gastrointestinal infections such as neonatal necrotizing enterocolitis. They are also administered topically to treat infections of the skin, mucous membranes, and ocular tissues.
Because of their highly ionized nature, aminoglycosides do not penetrate tissue cells significantly, and their volumes of distribution are similar to the extracellular fluid volume. Aminoglycosides also have poor penetration of the meninges, even when the meninges are inflamed, and intrathecal administration may be required to treat meningitis.
The aminoglycosides are not metabolized. They are excreted primarily by renal glomerular filtration, with little tubular reabsorption. The renal clearance of aminoglycosides is approximately equal to the creatinine clearance, because creatinine is also filtered at the glomerulus but is not secreted or reabsorbed significantly by the tubules. Because the clearance of aminoglycosides is proportional to the glomerular filtration rate, the dosage of aminoglycosides must be reduced in patients with renal impairment. In most cases, this is accomplished by increasing the interval between doses.
The plasma concentrations of aminoglycosides are routinely measured to ensure adequate dosage and to minimize toxicity. The peak concentration is found about 30 minutes after completing an intravenous infusion of an aminoglycoside, whereas the trough concentration is found immediately before administration of the next dose. Optimal peak and trough concentrations have been established and can be used to guide dosage adjustments for individuals receiving the standard regimen of three daily doses given at 8-hour intervals. For example, therapeutic concentrations of gentamicin and tobramycin are usually between 4 and 8 mg/L. A peak concentration above 12 mg/L or a trough concentration above 2 mg/L is considered toxic and indicates the need to reduce the dosage of gentamicin or tobramycin. Therapeutic concentrations of amikacin are between 16 and 32 mg/L, and the toxic peak and trough concentrations are above 35 mg/L and above 10 mg/L, respectively.
SPECTRUM AND INDICATIONS
The aminoglycosides are highly active against a wide range of aerobic gram-negative bacilli, and these antibiotics are now the most commonly used agents worldwide in the treatment of gram-negative bacterial infections. Gram-negative organisms have become increasingly resistant to β-lactam antibiotics and fluoroquinolones. Consequently, aminoglycosides have undergone resurgence in use.
Of the five aminoglycosides listed in Table 39–2, streptomycin is the least toxic, but it is also the least active against most gram-negative bacilli. Streptomycin is primarily used to treat tuberculosis and infections caused by Yersinia pestis (plague) and Francisella tularensis (tularemia).
Tobramycin is the most active aminoglycoside against many strains of P. aeruginosa, whereas gentamicin is usually more active against members of the family Enterobacteriaceae (Escherichia coli, Klebsiella spp. and others). Gentamicin is also used in combination with a penicillin to treat serious enterococcal, staphylococcal, or viridans group streptococcal infections. Amikacin is more resistant to bacterial enzymes that inactivate aminoglycosides, and it is active against some strains resistant to gentamicin and tobramycin.
BACTERIAL RESISTANCE
Resistance to aminoglycosides is primarily caused by inactivation of the drugs by bacterial enzymes that conjugate the drugs with acetate, phosphate, or adenylate. Resistance to aminoglycosides can also be caused by decreased binding of the drugs to the 30S ribosomal subunit or to decreased uptake of the drugs by porins in bacterial membranes (Table 39–3). Both plasmid and chromosomal genes are involved in resistance to aminoglycosides.
TABLE 39–3 Bacterial Resistance to Protein Synthesis Inhibitors
| Mechanism | Examples |
|---|---|
| Inactivation of the drug by bacterial enzymes | Inactivation of aminoglycosides by acetylase, adenylase, and phosphorylase enzymes |
| Inactivation of chloramphenicol by acetyltransferase | |
| Decreased binding of the drug | Decreased binding of aminoglycosides to the 30S ribosomal subunit |
| Decreased binding of macrolides and clindamycin to the 50S ribosomal subunit | |
| Decreased accumulation of the drug by bacteria | Active removal of macrolides from bacteria via membrane proteins |
| Decreased uptake of aminoglycosides via porins in bacterial membranes | |
| Decreased uptake of tetracyclines via porins in bacterial membranes |
ADVERSE EFFECTS
The most serious adverse effects of aminoglycosides are nephrotoxicity and ototoxicity. The risk of toxicity is related to the dosage and duration of treatment and varies with the specific drug. Irreversible toxicity can occur, even after use of the drug is discontinued, but serious toxicity is less likely if the offending drug is discontinued at the earliest sign of dysfunction.
Aminoglycosides are the most common cause of drug-induced renal failure. When the drugs accumulate in proximal tubule cells, they can cause acute tubular necrosis. Aminoglycosides also cause glomerular toxicity. These effects impair renal function and lead to a rise in plasma concentrations of the aminoglycosides. The elevated drug concentrations can further impair renal function and contribute to ototoxicity. Hence, it is important to monitor renal function and plasma aminoglycoside concentrations and to adjust the aminoglycoside dosage accordingly. Increasing the interval between doses to 24 hours or longer in persons with impaired renal function decreases the likelihood of toxicity.
Ototoxicity is associated with the accumulation of aminoglycosides in the labyrinth and hair cells of the cochlea, and has been attributed to activation of caspase-dependent apoptosis (programmed cell death) in hair cells. Patients can experience both vestibular and cochlear toxicity. Manifestations of vestibular toxicity include dizziness, impaired vision, nystagmus, vertigo, nausea, vomiting, and problems with postural balance and walking. Cochlear toxicity is characterized by tinnitus and hearing impairment and can lead to irreversible deafness. Often, a delay occurs between drug administration and the onset of symptoms, so many hospitalized patients are ambulatory before signs of toxicity appear.
The aminoglycosides vary in their tendency to cause cochlear or vestibular toxicity. Amikacin produces more cochlear toxicity (deafness), whereas gentamicin and streptomycin cause more vestibular toxicity. Tobramycin appears to cause similar degrees of cochlear and vestibular toxicity.
Neomycin is the most nephrotoxic aminoglycoside, and its use is limited to topical treatment of superficial infections. Neomycin is available in ointments and creams in combination with bacitracin and polymyxin. These triple-antibiotic preparations have been shown to prevent infections after minor skin trauma. Bacitracin provides gram-positive coverage, polymyxin provides gram-negative coverage, and neomycin is active against both gram-positive and gram-negative organisms. Neomycin can elicit hypersensitivity reactions, especially with long-term administration, and products containing only bacitracin and polymyxin are also available.
Aminocyclitol
Spectinomycin has an aminocyclitol structure that is similar to the structure of aminoglycoside drugs. As with the aminoglycosides, spectinomycin acts at the 30S ribosomal subunit. Spectinomycin is an alternative to ceftriaxone to treat gonorrhea caused by penicillinase-producing gonococci. Spectinomycin can cause nausea and produce pain at the site of injection.
Tetracyclines
CHEMISTRY AND PHARMACOKINETICS
The tetracycline antibiotics are four-ring anthracycline compounds produced by Streptomyces species. Doxycycline, minocycline, and tetracycline are semisynthetic derivatives of older tetracyclines, whereas tigecycline is a semisynthetic glycylcycline compound. The properties and clinical uses of these drugs are outlined in Tables 39–1 and 39–2.
The oral bioavailability of the tetracyclines varies from 70% for tetracycline to over 90% for doxycycline and minocycline. All tetracyclines bind divalent and trivalent cations, including calcium, aluminum, and iron. For this reason, their oral bioavailability is reduced if they are taken with foods or drugs containing these ions. Dairy products reduce the oral bioavailability of tetracycline but have little effect on the bioavailability of doxycycline and minocycline. None of the tetracyclines, however, should be taken with antacids or iron supplements.
The tetracycline drugs undergo minimal biotransformation and are excreted primarily in the urine and feces. Unlike other tetracyclines, doxycycline is not dependent on renal elimination, and doses do not need to be adjusted in persons with renal insufficiency.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


