FIGURE 164-3 Coronal fluid-attenuated inversion recovery (FLAIR) magnetic resonance image from a patient with herpes simplex encephalitis. Note the area of increased signal in the right temporal lobe (left side of image) confined predominantly to the gray matter. This patient had predominantly unilateral disease; bilateral lesions are more common but may be quite asymmetric in their intensity.
Significant MRI abnormalities are found in only approximately two-thirds of patients with WNV encephalitis, a frequency less than that with HSV encephalitis. When present, abnormalities often involve deep brain structures, including the thalamus, basal ganglia, and brainstem, rather than the cortex and may only be apparent on FLAIR images. EEGs in patients with WNV encephalitis typically show generalized slowing that may be more anteriorly prominent rather than the temporally predominant pattern of sharp or periodic discharges more characteristic of HSV encephalitis. Patients with VZV encephalitis may show multifocal areas of hemorrhagic and ischemic infarction, reflecting the tendency of this virus to produce a CNS vasculopathy rather than a true encephalitis. Immunocompromised adult patients with CMV often have enlarged ventricles with areas of increased T2 signal on MRI outlining the ventricles and subependymal enhancement on T1-weighted postcontrast images. Table 164-5 highlights specific diagnostic test results in encephalitis that can be useful in clinical decision making.
USE OF DIAGNOSTIC TESTS IN ENCEPHALITIS |
Abbreviations: CNS, central nervous system; CSF, cerebrospinal fluid; DWI, diffusion-weighted imaging; EA, early antigen; EBNA, EBV-associated nuclear antigen; EBV, Epstein-Barr virus; FLAIR, fluid-attenuated inversion recovery; HSV, herpes simplex virus; IgM, immunoglobulin M; MRI, magnetic resonance imaging; PCR, polymerase chain reaction; VCA, viral capsid antibody; VZV, varicella-zoster virus; WNV, West Nile virus.
Brain Biopsy Brain biopsy is now generally reserved for patients in whom CSF PCR studies fail to lead to a specific diagnosis, who have focal abnormalities on MRI, and who continue to show progressive clinical deterioration despite treatment with acyclovir and supportive therapy.
DIFFERENTIAL DIAGNOSIS
Infection by a variety of other organisms can mimic viral encephalitis. In studies of biopsy-proven HSV encephalitis, common infectious mimics of focal viral encephalitis included mycobacteria, fungi, rickettsiae, Listeria, Mycoplasma, and other bacteria (including Bartonella sp.). Autoimmune causes of encephalitis, including those associated with antibodies against N-methyl-D-aspartate (NMDA) receptor, voltage-gated potassium channels (VGKC), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and γ-aminobutyric acid (GABA) receptors, and GAD-65, have been increasingly recognized as causes of encephalitis that can mimic that caused by viral infection. In most cases, diagnosis is made by detection of the specific autoantibodies in serum and/or CSF. NMDA receptor antibodies have recently been reported in some patients with HSE encephalitis, and their presence should not exclude appropriate testing and treatment for HSV encephalitis. Autoimmune encephalitis may also be associated with specific cancers (paraneoplastic) and onconeuronal antibodies (e.g., anti-Hu, Yo, Ma2, amphiphysin, CRMP5, CV2) (Chap. 122). Subacute or chronic forms of encephalitis may occur in association with autoantibodies against thyroglobulin and thyroperoxidase (Hashimoto’s encephalopathy) and with prion diseases.
Infection caused by the ameba Naegleria fowleri can also cause acute meningoencephalitis (primary amebic meningoencephalitis), whereas that caused by Acanthamoeba and Balamuthia more typically produces subacute or chronic granulomatous amebic meningoencephalitis. Naegleria thrive in warm, iron-rich pools of water, including those found in drains, canals, and both natural and human-made outdoor pools. Infection has typically occurred in immunocompetent children with a history of swimming in potentially infected water. The CSF, in contrast to the typical profile seen in viral encephalitis, often resembles that of bacterial meningitis with a neutrophilic pleocytosis and hypoglycorrhachia. Motile trophozoites can be seen in a wet mount of warm, fresh CSF. There have been an increasing number of cases of Balamuthia mandrillaris amebic encephalitis mimicking acute viral encephalitis in children and immunocompetent adults. This organism has also been associated with encephalitis in recipients of transplanted organs from a donor with unrecognized infection. No effective treatment has been identified, and mortality approaches 100%.
Encephalitis can be caused by the raccoon pinworm Baylisascaris procyonis. Clues to the diagnosis include a history of raccoon exposure, especially of playing in or eating dirt potentially contaminated with raccoon feces. Most patients are children, and many have an associated eosinophilia.
Once nonviral causes of encephalitis have been excluded, the major diagnostic challenge is to distinguish HSV from other viruses that cause encephalitis. This distinction is particularly important because in virtually every other instance the therapy is supportive, whereas specific and effective antiviral therapy is available for HSV, and its efficacy is enhanced when it is instituted early in the course of infection. HSV encephalitis should be considered when clinical features suggesting involvement of the inferomedial frontotemporal regions of the brain are present, including prominent olfactory or gustatory hallucinations, anosmia, unusual or bizarre behavior or personality alterations, or memory disturbance. HSV encephalitis should always be suspected in patients with signs and symptoms consistent with acute encephalitis with focal findings on clinical examination, neuroimaging studies, or EEG. The diagnostic procedure of choice in these patients is CSF PCR analysis for HSV. A positive CSF PCR establishes the diagnosis, and a negative test dramatically reduces the likelihood of HSV encephalitis (see above).
![]() The anatomic distribution of lesions may provide an additional clue to diagnosis. Patients with rapidly progressive encephalitis and prominent brainstem signs, symptoms, or neuroimaging abnormalities may be infected by flaviviruses (WNV, St. Louis encephalitis virus, Japanese encephalitis virus), HSV, rabies, or L. monocytogenes. Significant involvement of deep gray matter structures, including the basal ganglia and thalamus, should also suggest possible flavivirus infection. These patients may present clinically with prominent movement disorders (tremor, myoclonus) or parkinsonian features. Patients with WNV infection can also present with a poliomyelitis-like acute flaccid paralysis, as can patients infected with EV71 and, less commonly, other enteroviruses. Acute flaccid paralysis is characterized by the acute onset of a lower motor neuron type of weakness with flaccid tone, reduced or absent reflexes, and relatively preserved sensation. The complete eradication of polio remains an ongoing challenge despite a continuing World Health Organization poliovirus elimination campaign. Three hundred forty-one cases of polio (almost all due to serotype 1) have been reported in 2013 from eight countries (Somalia 183 cases, Pakistan 63, Nigeria 51, Kenya 14, Syria 13, Afghanistan 9, Ethiopia 6, and Cameroon 2). There have been small outbreaks of poliomyelitis associated with vaccine strains of virus that have reverted to virulence through mutation or recombination with circulating wild-type enteroviruses in Hispaniola, China, the Philippines, Indonesia, Nigeria, and Madagascar.
The anatomic distribution of lesions may provide an additional clue to diagnosis. Patients with rapidly progressive encephalitis and prominent brainstem signs, symptoms, or neuroimaging abnormalities may be infected by flaviviruses (WNV, St. Louis encephalitis virus, Japanese encephalitis virus), HSV, rabies, or L. monocytogenes. Significant involvement of deep gray matter structures, including the basal ganglia and thalamus, should also suggest possible flavivirus infection. These patients may present clinically with prominent movement disorders (tremor, myoclonus) or parkinsonian features. Patients with WNV infection can also present with a poliomyelitis-like acute flaccid paralysis, as can patients infected with EV71 and, less commonly, other enteroviruses. Acute flaccid paralysis is characterized by the acute onset of a lower motor neuron type of weakness with flaccid tone, reduced or absent reflexes, and relatively preserved sensation. The complete eradication of polio remains an ongoing challenge despite a continuing World Health Organization poliovirus elimination campaign. Three hundred forty-one cases of polio (almost all due to serotype 1) have been reported in 2013 from eight countries (Somalia 183 cases, Pakistan 63, Nigeria 51, Kenya 14, Syria 13, Afghanistan 9, Ethiopia 6, and Cameroon 2). There have been small outbreaks of poliomyelitis associated with vaccine strains of virus that have reverted to virulence through mutation or recombination with circulating wild-type enteroviruses in Hispaniola, China, the Philippines, Indonesia, Nigeria, and Madagascar.
Epidemiologic factors may provide important clues to the diagnosis of viral meningitis or encephalitis. Particular attention should be paid to the season of the year; the geographic location and travel history; and possible exposure to animal bites or scratches, rodents, and ticks. Although transmission from the bite of an infected dog remains the most common cause of rabies worldwide, in the United States very few cases of dog rabies occur, and the most common risk factor is exposure to bats—although a clear history of a bite or scratch is often lacking. The classic clinical presentation of encephalitic (furious) rabies is fever, fluctuating consciousness, and autonomic hyperactivity. Phobic spasms of the larynx, pharynx, neck muscles, and diaphragm can be triggered by attempts to swallow water (hydrophobia) or by inspiration (aerophobia). Patients may also present with paralytic (dumb) rabies characterized by acute ascending paralysis. Rabies due to the bite of a bat has a different clinical presentation than classic rabies due to a dog or wolf bite. Patients present with focal neurologic deficits, myoclonus, seizures, and hallucinations; phobic spasms are not a typical feature. Patients with rabies have a CSF lymphocytic pleocytosis and may show areas of increased T2 signal abnormality in the brainstem, hippocampus, and hypothalamus. Diagnosis can be made by finding rabies virus antigen in brain tissue or in the neural innervation of hair follicles at the nape of the neck. PCR amplification of viral nucleic acid from CSF and saliva or tears may also enable diagnosis. Serology is frequently negative in both serum and CSF in the first week after onset of infection, which limits its acute diagnostic utility. No specific therapy is available, and cases are almost invariably fatal, with isolated survivors having devastating neurologic sequelae.
State public health authorities provide a valuable resource concerning isolation of particular agents in individual regions. Regular updates concerning the number, type, and distribution of cases of arboviral encephalitis can be found on the CDC and U.S. Geological Survey (USGS) websites (http://www.cdc.gov and http://diseasemaps.usgs.gov).
TREATMENT | VIRAL ENCEPHALITIS |
Specific antiviral therapy should be initiated when appropriate. Vital functions, including respiration and blood pressure, should be monitored continuously and supported as required. In the initial stages of encephalitis, many patients will require care in an intensive care unit. Basic management and supportive therapy should include careful monitoring of ICP, fluid restriction, avoidance of hypotonic intravenous solutions, and suppression of fever. Seizures should be treated with standard anticonvulsant regimens, and prophylactic therapy should be considered in view of the high frequency of seizures in severe cases of encephalitis. As with all seriously ill, immobilized patients with altered levels of consciousness, encephalitis patients are at risk for aspiration pneumonia, stasis ulcers and decubiti, contractures, deep venous thrombosis and its complications, and infections of indwelling lines and catheters.
Acyclovir is of benefit in the treatment of HSV and should be started empirically in patients with suspected viral encephalitis, especially if focal features are present, while awaiting viral diagnostic studies. Treatment should be discontinued in patients found not to have HSV encephalitis, with the possible exception of patients with severe encephalitis due to VZV or EBV. HSV, VZV, and EBV all encode an enzyme, deoxypyrimidine (thymidine) kinase, that phosphorylates acyclovir to produce acyclovir-5’-monophosphate. Host cell enzymes then phosphorylate this compound to form a triphosphate derivative. It is the triphosphate that acts as an antiviral agent by inhibiting viral DNA polymerase and by causing premature termination of nascent viral DNA chains. The specificity of action depends on the fact that uninfected cells do not phosphorylate significant amounts of acyclovir to acyclovir-5’-monophosphate. A second level of specificity is provided by the fact that the acyclovir triphosphate is a more potent inhibitor of viral DNA polymerase than of the analogous host cell enzymes.
Adults should receive a dose of 10 mg/kg of acyclovir intravenously every 8 h (30 mg/kg per day total dose) for 14–21 days. CSF PCR can be repeated at the completion of this course, with PCR-positive patients receiving additional treatment, followed by a repeat CSF PCR test. Neonatal HSV CNS infection is less responsive to acyclovir therapy than HSV encephalitis in adults; it is recommended that neonates with HSV encephalitis receive 20 mg/kg of acyclovir every 8 h (60 mg/kg per day total dose) for a minimum of 21 days.
Prior to intravenous administration, acyclovir should be diluted to a concentration ≤7 mg/mL. (A 70-kg person would receive a dose of 700 mg, which would be diluted in a volume of 100 mL.) Each dose should be infused slowly over 1 h, rather than by rapid or bolus infusion, to minimize the risk of renal dysfunction. Care should be taken to avoid extravasation or intramuscular or subcutaneous administration. The alkaline pH of acyclovir can cause local inflammation and phlebitis (9%). Dose adjustment is required in patients with impaired renal glomerular filtration. Penetration into CSF is excellent, with average drug levels ~50% of serum levels. Complications of therapy include elevations in blood urea nitrogen and creatinine levels (5%), thrombocytopenia (6%), gastrointestinal toxicity (nausea, vomiting, diarrhea) (7%), and neurotoxicity (lethargy or obtundation, disorientation, confusion, agitation, hallucinations, tremors, seizures) (1%). Acyclovir resistance may be mediated by changes in either the viral deoxypyrimidine kinase or DNA polymerase. To date, acyclovir-resistant isolates have not been a significant clinical problem in immunocompetent individuals. However, there have been reports of clinically virulent acyclovir-resistant HSV isolates from sites outside the CNS in immunocompromised individuals, including those with AIDS.
Oral antiviral drugs with efficacy against HSV, VZV, and EBV, including acyclovir, famciclovir, and valacyclovir, have not been evaluated in the treatment of encephalitis either as primary therapy or as supplemental therapy following completion of a course of parenteral acyclovir. A recently completed National Institute of Allergy and Infectious Disease (NIAID)/National Institute of Neurological Disorders and Stroke–sponsored phase III trial of supplemental oral valacyclovir therapy (2 g tid for 3 months) following the initial 14- to 21-day course of therapy with parenteral acyclovir (www.clinicaltrials.gov, identifier NCT00031486) was terminated early due to low enrollment. Although analysis was compromised due to low numbers, no differences were seen in the 12-month endpoints including dementia rating scale, mini-mental state exam, and Glasgow coma score in patients receiving valacyclovir versus placebo. The role of adjunctive intravenous glucocorticoids in treatment of HSV and VZV infection remains unclear, with most guidelines considering the existing supportive evidence weak and recommendation for possible use based on expert opinion only.
Ganciclovir and foscarnet, either alone or in combination, are often used in the treatment of CMV-related CNS infections, although their efficacy remains unproven. Cidofovir (see below) may provide an alternative in patients who fail to respond to ganciclovir and foscarnet, although data concerning its use in CMV CNS infections are extremely limited.
Ganciclovir is a synthetic nucleoside analogue of 2’-deoxyguanosine. The drug is preferentially phosphorylated by virus-induced cellular kinases. Ganciclovir triphosphate acts as a competitive inhibitor of the CMV DNA polymerase, and its incorporation into nascent viral DNA results in premature chain termination. Following intravenous administration, CSF concentrations of ganciclovir are 25–70% of coincident plasma levels. The usual dose for treatment of severe neurologic illnesses is 5 mg/kg every 12 h given intravenously at a constant rate over 1 h. Induction therapy is followed by maintenance therapy of 5 mg/kg every day for an indefinite period. Induction therapy should be continued until patients show a decline in CSF pleocytosis and a reduction in CSF CMV DNA copy number on quantitative PCR testing (where available). Doses should be adjusted in patients with renal insufficiency. Treatment is often limited by the development of granulocytopenia and thrombocytopenia (20–25%), which may require reduction in or discontinuation of therapy. Gastrointestinal side effects, including nausea, vomiting, diarrhea, and abdominal pain, occur in ~20% of patients. Some patients treated with ganciclovir for CMV retinitis have developed retinal detachment, but the causal relationship to ganciclovir treatment is unclear. Valganciclovir is an orally bioavailable prodrug that can generate high serum levels of ganciclovir, although studies of its efficacy in treating CMV CNS infections are limited.
Foscarnet is a pyrophosphate analogue that inhibits viral DNA polymerases by binding to the pyrophosphate-binding site. Following intravenous infusion, CSF concentrations range from 15 to 100% of coincident plasma levels. The usual dose for serious CMV-related neurologic illness is 60 mg/kg every 8 h administered by constant infusion over 1 h. Induction therapy for 14–21 days is followed by maintenance therapy (60–120 mg/kg per day). Induction therapy may need to be extended in patients who fail to show a decline in CSF pleocytosis and a reduction in CSF CMV DNA copy number on quantitative PCR tests (where available). Approximately one-third of patients develop renal impairment during treatment, which is reversible following discontinuation of therapy in most, but not all, cases. This is often associated with elevations in serum creatinine and proteinuria and is less frequent in patients who are adequately hydrated. Many patients experience fatigue and nausea. Reductions in serum calcium, magnesium, and potassium occur in ~15% of patients and may be associated with tetany, cardiac rhythm disturbances, or seizures.
Cidofovir is a nucleotide analogue that is effective in treating CMV retinitis and equivalent to or better than ganciclovir in some experimental models of murine CMV encephalitis, although data concerning its efficacy in human CMV CNS disease are limited. The usual dose is 5 mg/kg intravenously once weekly for 2 weeks, then biweekly for two or more additional doses, depending on clinical response. Patients must be prehydrated with normal saline (e.g., 1 L over 1–2 h) prior to each dose and treated with probenecid (e.g., 1 g 3 h before cidofovir and 1 g 2 and 8 h after cidofovir). Nephrotoxicity is common; the dose should be reduced if renal function deteriorates.
Intravenous ribavirin (15–25 mg/kg per day in divided doses given every 8 h) has been reported to be of benefit in isolated cases of severe encephalitis due to California encephalitis (La Crosse) virus. Ribavirin might be of benefit for the rare patients, typically infants or young children, with severe adenovirus or rotavirus encephalitis and in patients with encephalitis due to LCMV or other arenaviruses. However, clinical trials are lacking. Hemolysis, with resulting anemia, has been the major side effect limiting therapy.
No specific antiviral therapy of proven efficacy is currently available for treatment of WNV encephalitis. Patients have been treated with a-interferon, ribavirin, an Israeli IVIg preparation that contains high-titer anti-WNV antibody (Omr-IgG-am) (www.clinicaltrials.gov, identifier NCT00069316 and 0068055), and humanized monoclonal antibodies directed against the viral envelope glycoprotein (www.clinicaltrials.gov, identifier NCT00927953 and 00515385). WNV chimeric vaccines, in which WNV envelope and premembrane proteins are inserted into the background of another flavivirus, are already undergoing human clinical testing and have been found to be both safe and immunogenic in healthy adults but have not yet been tested for disease prevention in humans (www.clinicaltrials.gov, identifier NCT00746798, 00442169, 00094718, and 00537147). Both chimeric and killed inactivated WNV vaccines have been found to be safe and effective in preventing equine WNV infection, and several effective flavivirus vaccines are already in human use, creating optimism that a safe and effective human WNV vaccine can also be developed.
SEQUELAE
There is considerable variation in the incidence and severity of sequelae in patients surviving viral encephalitis. In the case of EEE virus infection, nearly 80% of survivors have severe neurologic sequelae. At the other extreme are infections due to EBV, California encephalitis virus, and Venezuelan equine encephalitis virus, where severe sequelae are unusual. For example, approximately 5–15% of children infected with La Crosse virus have a residual seizure disorder, and 1% have persistent hemiparesis. Detailed information about sequelae in patients with HSV encephalitis treated with acyclovir is available from the NIAID-Collaborative Antiviral Study Group (CASG) trials. Of 32 acyclovir-treated patients, 26 survived (81%). Of the 26 survivors, 12 (46%) had no or only minor sequelae, 3 (12%) were moderately impaired (gainfully employed but not functioning at their previous level), and 11 (42%) were severely impaired (requiring continuous supportive care). The incidence and severity of sequelae were directly related to the age of the patient and the level of consciousness at the time of initiation of therapy. Patients with severe neurologic impairment (Glasgow coma score 6) at initiation of therapy either died or survived with severe sequelae. Young patients (<30 years) with good neurologic function at initiation of therapy did substantially better (100% survival, 62% with no or mild sequelae) compared with their older counterparts (>30 years; 64% survival, 57% no or mild sequelae). Some recent studies using quantitative HSV CSF PCR tests indicate that clinical outcome following treatment also correlates with the amount of HSV DNA present in CSF at the time of presentation. Many patients with WNV infection have sequelae, including cognitive impairment; weakness; and hyper- or hypokinetic movement disorders, including tremor, myoclonus, and parkinsonism. In a large longitudinal study of prognosis in 156 patients with WNV infection, the mean time to achieve recovery (defined as 95% of maximal predicted score on specific validated tests) was 112–148 days for fatigue, 121–175 days for physical function, 131–139 days for mood, and 302–455 days for mental function (the longer interval in each case representing patients with neuroinvasive disease).
SUBACUTE MENINGITIS
CLINICAL MANIFESTATIONS
Patients with subacute meningitis typically have an unrelenting headache, stiff neck, low-grade fever, and lethargy for days to several weeks before they present for evaluation. Cranial nerve abnormalities and night sweats may be present. This syndrome overlaps that of chronic meningitis, discussed in detail in Chap. 165.
ETIOLOGY
Common causative organisms include M. tuberculosis, C. neoformans, H. capsulatum, C. immitis, and T. pallidum. Initial infection with M. tuberculosis is acquired by inhalation of aerosolized droplet nuclei. Tuberculous meningitis in adults does not develop acutely from hematogenous spread of tubercle bacilli to the meninges. Rather, millet seed–sized (miliary) tubercles form in the parenchyma of the brain during hematogenous dissemination of tubercle bacilli in the course of primary infection. These tubercles enlarge and are usually caseating. The propensity for a caseous lesion to produce meningitis is determined by its proximity to the subarachnoid space (SAS) and the rate at which fibrous encapsulation develops. Subependymal caseous foci cause meningitis via discharge of bacilli and tuberculous antigens into the SAS. Mycobacterial antigens produce an intense inflammatory reaction that leads to the production of a thick exudate that fills the basilar cisterns and surrounds the cranial nerves and major blood vessels at the base of the brain.
![]() Fungal infections are typically acquired by the inhalation of airborne fungal spores. The initial pulmonary infection may be asymptomatic or present with fever, cough, sputum production, and chest pain. The pulmonary infection is often self-limited. A localized pulmonary fungal infection can then remain dormant in the lungs until there is an abnormality in cell-mediated immunity that allows the fungus to reactivate and disseminate to the CNS. The most common pathogen causing fungal meningitis is C. neoformans. This fungus is found worldwide in soil and bird excreta. H. capsulatum is endemic to the Ohio and Mississippi River valleys of the central United States and to parts of Central and South America. C. immitis is endemic to the desert areas of the southwest United States, northern Mexico, and Argentina.
Fungal infections are typically acquired by the inhalation of airborne fungal spores. The initial pulmonary infection may be asymptomatic or present with fever, cough, sputum production, and chest pain. The pulmonary infection is often self-limited. A localized pulmonary fungal infection can then remain dormant in the lungs until there is an abnormality in cell-mediated immunity that allows the fungus to reactivate and disseminate to the CNS. The most common pathogen causing fungal meningitis is C. neoformans. This fungus is found worldwide in soil and bird excreta. H. capsulatum is endemic to the Ohio and Mississippi River valleys of the central United States and to parts of Central and South America. C. immitis is endemic to the desert areas of the southwest United States, northern Mexico, and Argentina.
Syphilis is a sexually transmitted disease that is manifest by the appearance of a painless chancre at the site of inoculation. T. pallidum invades the CNS early in the course of syphilis. Cranial nerves VII and VIII are most frequently involved.
LABORATORY DIAGNOSIS
The classic CSF abnormalities in tuberculous meningitis are as follows: (1) elevated opening pressure, (2) lymphocytic pleocytosis (10–500 cells/μL), (3) elevated protein concentration in the range of 1–5 g/L, and (4) decreased glucose concentration in the range of 1.1–2.2 mmol/L (20–40 mg/dL). The combination of unrelenting headache, stiff neck, fatigue, night sweats, and fever with a CSF lymphocytic pleocytosis and a mildly decreased glucose concentration is highly suspicious for tuberculous meningitis. The last tube of fluid collected at LP is the best tube to send for a smear for acid-fast bacilli (AFB). If there is a pellicle in the CSF or a cobweb-like clot on the surface of the fluid, AFB can best be demonstrated in a smear of the clot or pellicle. Positive smears are typically reported in only 10–40% of cases of tuberculous meningitis in adults. Cultures of CSF take 4–8 weeks to identify the organism and are positive in ~50% of adults. Culture remains the gold standard to make the diagnosis of tuberculous meningitis. PCR for the detection of M. tuberculosis DNA should be sent on CSF if available, but the sensitivity and specificity on CSF have not been defined. The CDC recommends the use of nucleic acid amplification tests for the diagnosis of pulmonary tuberculosis.
The characteristic CSF abnormalities in fungal meningitis are a mononuclear or lymphocytic pleocytosis, an increased protein concentration, and a decreased glucose concentration. There may be eosinophils in the CSF in C. immitis meningitis. Large volumes of CSF are often required to demonstrate the organism on India ink smear or grow the organism in culture. If spinal fluid examined by LP on two separate occasions fails to yield an organism, CSF should be obtained by high-cervical or cisternal puncture.
The cryptococcal polysaccharide antigen test is a highly sensitive and specific test for cryptococcal meningitis. A reactive CSF cryptococcal antigen test establishes the diagnosis. The detection of the Histoplasma polysaccharide antigen in CSF establishes the diagnosis of a fungal meningitis but is not specific for meningitis due to H. capsulatum. It may be falsely positive in coccidioidal meningitis. The CSF complement fixation antibody test is reported to have a specificity of 100% and a sensitivity of 75% for coccidioidal meningitis.
The diagnosis of syphilitic meningitis is made when a reactive serum treponemal test (fluorescent treponemal antibody absorption test [FTA-ABS] or microhemagglutination assay–T. pallidum [MHA-TP]) is associated with a CSF lymphocytic or mononuclear pleocytosis and an elevated protein concentration, or when the CSF Venereal Disease Research Laboratory (VDRL) test is positive. A reactive CSF FTA-ABS is not definitive evidence of neurosyphilis. The CSF FTA-ABS can be falsely positive from blood contamination. A negative CSF VDRL does not rule out neurosyphilis. A negative CSF FTA-ABS or MHA-TP rules out neurosyphilis.
TREATMENT | SUBACUTE MENINGITIS |
Empirical therapy of tuberculous meningitis is often initiated on the basis of a high index of suspicion without adequate laboratory support. Initial therapy is a combination of isoniazid (300 mg/d), rifampin (10 mg/kg per day), pyrazinamide (30 mg/kg per day in divided doses), ethambutol (15–25 mg/kg per day in divided doses), and pyridoxine (50 mg/d). When the antimicrobial sensitivity of the M. tuberculosis isolate is known, ethambutol can be discontinued. If the clinical response is good, pyrazinamide can be discontinued after 8 weeks and isoniazid and rifampin continued alone for the next 6–12 months. A 6-month course of therapy is acceptable, but therapy should be prolonged for 9–12 months in patients who have an inadequate resolution of symptoms of meningitis or who have positive mycobacterial cultures of CSF during the course of therapy. Dexamethasone therapy is recommended for HIV-negative patients with tuberculous meningitis. The dose is 12–16 mg/d for 3 weeks, and then tapered over 3 weeks.
Meningitis due to C. neoformans in non-HIV, nontransplant patients is treated with induction therapy with amphotericin B (AmB) (0.7 mg/kg IV per day) plus flucytosine (100 mg/kg per day in four divided doses) for at least 4 weeks if CSF culture results are negative after 2 weeks of treatment. Therapy should be extended for a total of 6 weeks in the patient with neurologic complications. Induction therapy is followed by consolidation therapy with fluconazole 400 mg/d for 8 weeks. Organ transplant recipients are treated with liposomal AmB (3–4 mg/kg per day) or AmB lipid complex (ABLC) 5 mg/kg per day plus flucytosine (100 mg/kg per day in four divided doses) for at least 2 weeks or until CSF culture is sterile. Follow CSF yeast cultures for sterilization rather than the cryptococcal antigen titer. This treatment is followed by an 8- to 10-week course of fluconazole (400–800 mg/d [6–12 mg/kg] PO). If the CSF culture is sterile after 10 weeks of acute therapy, the dose of fluconazole is decreased to 200 mg/d for 6 months to a year. Patients with HIV infection are treated with AmB or a lipid formulation plus flucytosine for at least 2 weeks, followed by fluconazole for a minimum of 8 weeks. HIV-infected patients may require indefinite maintenance therapy with fluconazole 200 mg/d. Meningitis due to H. capsulatum is treated with AmB (0.7–1.0 mg/kg per day) for 4–12 weeks. A total dose of 30 mg/kg is recommended. Therapy with AmB is not discontinued until fungal cultures are sterile. After completing a course of AmB, maintenance therapy with itraconazole 200 mg two or three times daily is initiated and continued for at least 9 months to a year. C. immitis meningitis is treated with either high-dose fluconazole (1000 mg daily) as monotherapy or intravenous AmB (0.5–0.7 mg/kg per day) for >4 weeks. Intrathecal AmB (0.25–0.75 mg/d three times weekly) may be required to eradicate the infection. Lifelong therapy with fluconazole (200–400 mg daily) is recommended to prevent relapse. AmBisome (5 mg/kg per day) or AmB lipid complex (5 mg/kg per day) can be substituted for AmB in patients who have or who develop significant renal dysfunction. The most common complication of fungal meningitis is hydrocephalus. Patients who develop hydrocephalus should receive a CSF diversion device. A ventriculostomy can be used until CSF fungal cultures are sterile, at which time the ventriculostomy is replaced by a ventriculoperitoneal shunt.
Syphilitic meningitis is treated with aqueous penicillin G in a dose of 3–4 million units intravenously every 4 h for 10–14 days. An alternative regimen is 2.4 million units of procaine penicillin G intramuscularly daily with 500 mg of oral probenecid four times daily for 10–14 days. Either regimen is followed with 2.4 million units of benzathine penicillin G intramuscularly once a week for 3 weeks. The standard criterion for treatment success is reexamination of the CSF. The CSF should be reexamined at 6-month intervals for 2 years. The cell count is expected to normalize within 12 months, and the VDRL titer to decrease by two dilutions or revert to nonreactive within 2 years of completion of therapy. Failure of the CSF pleocytosis to resolve or an increase in the CSF VDRL titer by two or more dilutions requires retreatment.
CHRONIC ENCEPHALITIS
PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
Clinical Features and Pathology Progressive multifocal leukoencephalopathy (PML) is characterized pathologically by multifocal areas of demyelination of varying size distributed throughout the brain but sparing the spinal cord and optic nerves. In addition to demyelination, there are characteristic cytologic alterations in both astrocytes and oligodendrocytes. Astrocytes are enlarged and contain hyperchromatic, deformed, and bizarre nuclei and frequent mitotic figures. Oligodendrocytes have enlarged, densely staining nuclei that contain viral inclusions formed by crystalline arrays of JC virus (JCV) particles. Patients often present with visual deficits (45%), typically a homonymous hemianopia; mental impairment (38%) (dementia, confusion, personality change); weakness, including hemi- or monoparesis; and ataxia. Seizures occur in ~20% of patients, predominantly in those with lesions abutting the cortex.
Almost all patients have an underlying immunosuppressive disorder or are receiving immunomodulatory therapy. In recent series, the most common associated conditions were AIDS (80%), hematologic malignancies (13%), transplant recipients (5%), and chronic inflammatory diseases (2%). It has been estimated that up to 5% of AIDS patients will develop PML. There have been over 400 reported cases of PML occurring in patients being treated for multiple sclerosis and inflammatory bowel disease with natalizumab, a humanized monoclonal antibody that inhibits lymphocyte trafficking into CNS and bowel mucosa by binding to α4 integrins. Overall risk in these patients has been estimated at ~3.4 PML cases per 1000 treated patients, but the risk depends on a variety of factors including anti-JCV antibody serostatus, prior immunosuppressive therapy use, and duration of natalizumab therapy. Patients who lack detectable JCV antibody have a risk of developing PML of <0.1 case/1000 patients, whereas those who are JCV seropositive and have been exposed to prior immunosuppressive therapy and have received >24 months of natalizumab therapy have a risk of >1 case/100 treated patients. PML cases have also been reported in patients receiving other humanized monoclonal antibodies with immunomodulatory activity including efalizumab and rituximab, although the relative risks have not been clearly established. The basic clinical and diagnostic features appear to be similar in HIV-associated PML and PML associated with immunomodulatory drugs with the exception of an increased likelihood of peripheral enhancement in MRIs of PML lesions in immunomodulatory cases. In natalizumab-associated PML, patients will also almost invariably develop clinical and radiographic worsening of lesions with discontinuation of therapy, attributed to development of immune reconstitution inflammatory syndrome (IRIS).
Diagnostic Studies The diagnosis of PML is frequently suggested by MRI. MRI reveals multifocal asymmetric, coalescing white matter lesions located periventricularly, in the centrum semiovale, in the parietal-occipital region, and in the cerebellum. These lesions have increased signal on T2 and FLAIR images and decreased signal on T1-weighted images. HIV-PML lesions are classically nonenhancing (90%), but patients with immunomodulatory drug associated PML may have peripheral ring enhancement. PML lesions are not typically associated with edema or mass effect. CT scans, which are less sensitive than MRI for the diagnosis of PML, often show hypodense nonenhancing white matter lesions.
The CSF is typically normal, although mild elevation in protein and/or IgG may be found. Pleocytosis occurs in <25% of cases, is predominantly mononuclear, and rarely exceeds 25 cells/μL. PCR amplification of JCV DNA from CSF has become an important diagnostic tool. The presence of a positive CSF PCR for JCV DNA in association with typical MRI lesions in the appropriate clinical setting is diagnostic of PML, reflecting the assay’s relatively high specificity (92–100%); however, sensitivity is variable, and a negative CSF PCR does not exclude the diagnosis. In HIV-negative patients and HIV-positive patients not receiving highly active antiviral therapy (HAART), sensitivity is likely 70–90%. In HAART-treated patients, sensitivity may be closer to 60%, reflecting the lower JCV CSF viral load in this relatively more immunocompetent group. Studies with quantitative JCV CSF PCR indicate that patients with low JCV loads (<100 copies/μL) have a generally better prognosis than those with higher viral loads. Patients with negative CSF PCR studies may require brain biopsy for definitive diagnosis. In biopsy or necropsy specimens of brain, JCV antigen and nucleic acid can be detected by immunocytochemistry, in situ hybridization, or PCR amplification.
Serologic studies are of no utility in diagnosis due to high basal seroprevalence level, but may contribute to risk stratification in patients contemplating therapy with immunomodulatory drugs such as natalizumab.
TREATMENT | PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY |
No effective therapy for PML is available. There are case reports of potential beneficial effects of the 5-HT2a receptor antagonist mirtazapine, which may inhibit binding of JCV to its receptor on oligodendrocytes. Retrospective noncontrolled studies have also suggested a possible beneficial effect of treatment with interferon-α. Neither of these agents has been tested in randomized controlled clinical trials. A prospective multicenter clinical trial to evaluate the efficacy of the antimalarial drug mefloquine failed to show benefit. Intravenous and/or intrathecal cytarabine were not shown to be of benefit in a randomized controlled trial in HIV-associated PML, although some experts suggest that cytarabine may have therapeutic efficacy in situations where breakdown of the blood-brain barrier allows sufficient CSF penetration. A randomized controlled trial of cidofovir in HIV-associated PML also failed to show significant benefit. Because PML almost invariably occurs in immunocompromised individuals, any therapeutic interventions designed to enhance or restore immunocompetence should be considered. Perhaps the most dramatic demonstration of this is disease stabilization and, in rare cases, improvement associated with the improvement in the immune status of HIV-positive patients with AIDS following institution of HAART. In HIV-positive PML patients treated with HAART, 1-year survival is ~50%, although up to 80% of survivors may have significant neurologic sequelae. HIV-positive PML patients with higher CD4 counts (>300/μL) and low or nondetectable HIV viral loads have a better prognosis than those with lower CD4 counts and higher viral loads. Although institution of HAART enhances survival in HIV-positive PML patients, the associated immune reconstitution in patients with an underlying opportunistic infection such as PML may also result in a severe CNS inflammatory syndrome (IRIS) associated with clinical worsening, CSF pleocytosis, and the appearance of new enhancing MRI lesions. Patients receiving natalizumab or other immunomodulatory antibodies, who are suspected of having PML, should have therapy immediately halted and circulating antibodies removed by plasma exchange. Patients should be closely monitored for development of IRIS, which is generally treated with intravenous glucocorticoids, although controlled clinical trials of efficacy remain lacking.
SUBACUTE SCLEROSING PANENCEPHALITIS (SSPE)
SSPE is a rare chronic, progressive demyelinating disease of the CNS associated with a chronic nonpermissive infection of brain tissue with measles virus. The frequency has been estimated at 1 in 100,000–500,000 measles cases. An average of five cases per year are reported in the United States. The incidence has declined dramatically since the introduction of a measles vaccine. Most patients give a history of primary measles infection at an early age (2 years), which is followed after a latent interval of 6–8 years by the development of a progressive neurologic disorder. Some 85% of patients are between 5 and 15 years old at diagnosis. Initial manifestations include poor school performance and mood and personality changes. Typical signs of a CNS viral infection, including fever and headache, do not occur. As the disease progresses, patients develop progressive intellectual deterioration, focal and/or generalized seizures, myoclonus, ataxia, and visual disturbances. In the late stage of the illness, patients are unresponsive, quadriparetic, and spastic, with hyperactive tendon reflexes and extensor plantar responses.
Diagnostic Studies MRI is often normal early, although areas of increased T2 signal develop in the white matter of the brain and brainstem as disease progresses. The EEG may initially show only nonspecific slowing, but with disease progression, patients develop a characteristic periodic pattern with bursts of high-voltage, sharp, slow waves every 3–8 s, followed by periods of attenuated (“flat”) background. The CSF is acellular with a normal or mildly elevated protein concentration and a markedly elevated gamma globulin level (>20% of total CSF protein). CSF antimeasles antibody levels are invariably elevated, and oligoclonal antimeasles antibodies are often present. Measles virus can be cultured from brain tissue using special cocultivation techniques. Viral antigen can be identified immunocytochemically, and viral genome can be detected by in situ hybridization or PCR amplification.
TREATMENT | SUBACUTE SCLEROSING PANENCEPHALITIS |
No definitive therapy for SSPE is available. Treatment with isoprinosine (Inosiplex, 100 mg/kg per day), alone or in combination with intrathecal or intraventricular interferon-α, has been reported to prolong survival and produce clinical improvement in some patients but has never been subjected to a controlled clinical trial.
PROGRESSIVE RUBELLA PANENCEPHALITIS
This is an extremely rare disorder that primarily affects males with congenital rubella syndrome, although isolated cases have been reported following childhood rubella. After a latent period of 8–19 years, patients develop progressive neurologic deterioration. The manifestations are similar to those seen in SSPE. CSF shows a mild lymphocytic pleocytosis, slightly elevated protein concentration, markedly increased gamma globulin, and rubella virus–specific oligoclonal bands. No therapy is available. Universal prevention of both congenital and childhood rubella through the use of the available live attenuated rubella vaccine would be expected to eliminate the disease.
BRAIN ABSCESS
DEFINITION
A brain abscess is a focal, suppurative infection within the brain parenchyma, typically surrounded by a vascularized capsule. The term cerebritis is often employed to describe a nonencapsulated brain abscess.
EPIDEMIOLOGY
![]() A bacterial brain abscess is a relatively uncommon intracranial infection, with an incidence of ~0.3–1.3:100,000 persons per year. Predisposing conditions include otitis media and mastoiditis, paranasal sinusitis, pyogenic infections in the chest or other body sites, penetrating head trauma or neurosurgical procedures, and dental infections. In immunocompetent individuals the most important pathogens are Streptococcus spp. (anaerobic, aerobic, and viridans [40%]), Enterobacteriaceae (Proteus spp., E. coli sp., Klebsiella spp. [25%]), anaerobes (e.g., Bacteroides spp., Fusobacterium spp. [30%]), and staphylococci (10%). In immunocompromised hosts with underlying HIV infection, organ transplantation, cancer, or immunosuppressive therapy, most brain abscesses are caused by Nocardia spp., Toxoplasma gondii, Aspergillus spp., Candida spp., and C. neoformans. In Latin America and in immigrants from Latin America, the most common cause of brain abscess is Taenia solium (neurocysticercosis). In India and East Asia, mycobacterial infection (tuberculoma) remains a major cause of focal CNS mass lesions.
A bacterial brain abscess is a relatively uncommon intracranial infection, with an incidence of ~0.3–1.3:100,000 persons per year. Predisposing conditions include otitis media and mastoiditis, paranasal sinusitis, pyogenic infections in the chest or other body sites, penetrating head trauma or neurosurgical procedures, and dental infections. In immunocompetent individuals the most important pathogens are Streptococcus spp. (anaerobic, aerobic, and viridans [40%]), Enterobacteriaceae (Proteus spp., E. coli sp., Klebsiella spp. [25%]), anaerobes (e.g., Bacteroides spp., Fusobacterium spp. [30%]), and staphylococci (10%). In immunocompromised hosts with underlying HIV infection, organ transplantation, cancer, or immunosuppressive therapy, most brain abscesses are caused by Nocardia spp., Toxoplasma gondii, Aspergillus spp., Candida spp., and C. neoformans. In Latin America and in immigrants from Latin America, the most common cause of brain abscess is Taenia solium (neurocysticercosis). In India and East Asia, mycobacterial infection (tuberculoma) remains a major cause of focal CNS mass lesions.
ETIOLOGY
A brain abscess may develop (1) by direct spread from a contiguous cranial site of infection, such as paranasal sinusitis, otitis media, mastoiditis, or dental infection; (2) following head trauma or a neurosurgical procedure; or (3) as a result of hematogenous spread from a remote site of infection. In up to 25% of cases, no obvious primary source of infection is apparent (cryptogenic brain abscess).
Approximately one-third of brain abscesses are associated with otitis media and mastoiditis, often with an associated cholesteatoma. Otogenic abscesses occur predominantly in the temporal lobe (55–75%) and cerebellum (20–30%). In some series, up to 90% of cerebellar abscesses are otogenic. Common organisms include streptococci, Bacteroides spp., Pseudomonas spp., Haemophilus spp., and Enterobacteriaceae. Abscesses that develop as a result of direct spread of infection from the frontal, ethmoidal, or sphenoidal sinuses and those that occur due to dental infections are usually located in the frontal lobes. Approximately 10% of brain abscesses are associated with paranasal sinusitis, and this association is particularly strong in young males in their second and third decades of life. The most common pathogens in brain abscesses associated with paranasal sinusitis are streptococci (especially Streptococcus milleri), Haemophilus spp., Bacteroides spp., Pseudomonas spp., and S. aureus. Dental infections are associated with ~2% of brain abscesses, although it is often suggested that many “cryptogenic” abscesses are in fact due to dental infections. The most common pathogens in this setting are streptococci, staphylococci, Bacteroides spp., and Fusobacterium spp.
Hematogenous abscesses account for ~25% of brain abscesses. Hematogenous abscesses are often multiple, and multiple abscesses often (50%) have a hematogenous origin. These abscesses show a predilection for the territory of the middle cerebral artery (i.e., posterior frontal or parietal lobes). Hematogenous abscesses are often located at the junction of the gray and white matter and are often poorly encapsulated. The microbiology of hematogenous abscesses is dependent on the primary source of infection. For example, brain abscesses that develop as a complication of infective endocarditis are often due to viridans streptococci or S. aureus. Abscesses associated with pyogenic lung infections such as lung abscess or bronchiectasis are often due to streptococci, staphylococci, Bacteroides spp., Fusobacterium spp., or Enterobacteriaceae. Abscesses that follow penetrating head trauma or neurosurgical procedures are frequently due to methicillin-resistant S. aureus (MRSA), S. epidermidis, Enterobacteriaceae, Pseudomonas spp., and Clostridium spp. Enterobacteriaceae and P. aeruginosa are important causes of abscesses associated with urinary sepsis. Congenital cardiac malformations that produce a right-to-left shunt, such as tetralogy of Fallot, patent ductus arteriosus, and atrial and ventricular septal defects, allow bloodborne bacteria to bypass the pulmonary capillary bed and reach the brain. Similar phenomena can occur with pulmonary arteriovenous malformations. The decreased arterial oxygenation and saturation from the right-to-left shunt and polycythemia may cause focal areas of cerebral ischemia, thus providing a nidus for microorganisms that bypassed the pulmonary circulation to multiply and form an abscess. Streptococci are the most common pathogens in this setting.
PATHOGENESIS AND HISTOPATHOLOGY
Results of experimental models of brain abscess formation suggest that for bacterial invasion of brain parenchyma to occur, there must be preexisting or concomitant areas of ischemia, necrosis, or hypoxemia in brain tissue. The intact brain parenchyma is relatively resistant to infection. Once bacteria have established infection, brain abscess frequently evolves through a series of stages, influenced by the nature of the infecting organism and by the immunocompetence of the host. The early cerebritis stage (days 1–3) is characterized by a perivascular infiltration of inflammatory cells, which surround a central core of coagulative necrosis. Marked edema surrounds the lesion at this stage. In the late cerebritis stage (days 4–9), pus formation leads to enlargement of the necrotic center, which is surrounded at its border by an inflammatory infiltrate of macrophages and fibroblasts. A thin capsule of fibroblasts and reticular fibers gradually develops, and the surrounding area of cerebral edema becomes more distinct than in the previous stage. The third stage, early capsule formation (days 10–13), is characterized by the formation of a capsule that is better developed on the cortical than on the ventricular side of the lesion. This stage correlates with the appearance of a ring-enhancing capsule on neuroimaging studies. The final stage, late capsule formation (day 14 and beyond), is defined by a well-formed necrotic center surrounded by a dense collagenous capsule. The surrounding area of cerebral edema has regressed, but marked gliosis with large numbers of reactive astrocytes has developed outside the capsule. This gliotic process may contribute to the development of seizures as a sequela of brain abscess.
CLINICAL PRESENTATION
A brain abscess typically presents as an expanding intracranial mass lesion rather than as an infectious process. Although the evolution of signs and symptoms is extremely variable, ranging from hours to weeks or even months, most patients present to the hospital 11–12 days following onset of symptoms. The classic clinical triad of headache, fever, and a focal neurologic deficit is present in <50% of cases. The most common symptom in patients with a brain abscess is headache, occurring in >75% of patients. The headache is often characterized as a constant, dull, aching sensation, either hemicranial or generalized, and it becomes progressively more severe and refractory to therapy. Fever is present in only 50% of patients at the time of diagnosis, and its absence should not exclude the diagnosis. The new onset of focal or generalized seizure activity is a presenting sign in 15–35% of patients. Focal neurologic deficits including hemiparesis, aphasia, or visual field defects are part of the initial presentation in >60% of patients.
The clinical presentation of a brain abscess depends on its location, the nature of the primary infection if present, and the level of the ICP. Hemiparesis is the most common localizing sign of a frontal lobe abscess. A temporal lobe abscess may present with a disturbance of language (dysphasia) or an upper homonymous quadrantanopia. Nystagmus and ataxia are signs of a cerebellar abscess. Signs of raised ICP—papilledema, nausea and vomiting, and drowsiness or confusion—can be the dominant presentation of some abscesses, particularly those in the cerebellum. Meningismus is not present unless the abscess has ruptured into the ventricle or the infection has spread to the subarachnoid space.
DIAGNOSIS
Diagnosis is made by neuroimaging studies. MRI (Fig. 164-4) is better than CT for demonstrating abscesses in the early (cerebritis) stages and is superior to CT for identifying abscesses in the posterior fossa. Cerebritis appears on MRI as an area of low-signal intensity on T1-weighted images with irregular postgadolinium enhancement and as an area of increased signal intensity on T2-weighted images. Cerebritis is often not visualized by CT scan, but when present, appears as an area of hypodensity. On a contrast-enhanced CT scan, a mature brain abscess appears as a focal area of hypodensity surrounded by ring enhancement with surrounding edema (hypodensity). On contrast-enhanced T1-weighted MRI, a mature brain abscess has a capsule that enhances surrounding a hypodense center and surrounded by a hypodense area of edema. On T2-weighted MRI, there is a hyperintense central area of pus surrounded by a well-defined hypointense capsule and a hyperintense surrounding area of edema. It is important to recognize that the CT and MRI appearance, particularly of the capsule, may be altered by treatment with glucocorticoids. The distinction between a brain abscess and other focal CNS lesions such as primary or metastatic tumors may be facilitated by the use of diffusion-weighted imaging sequences on which a brain abscess typically shows increased signal due to restricted diffusion of the abscess cavity with corresponding low signal on apparent diffusion coefficient images.
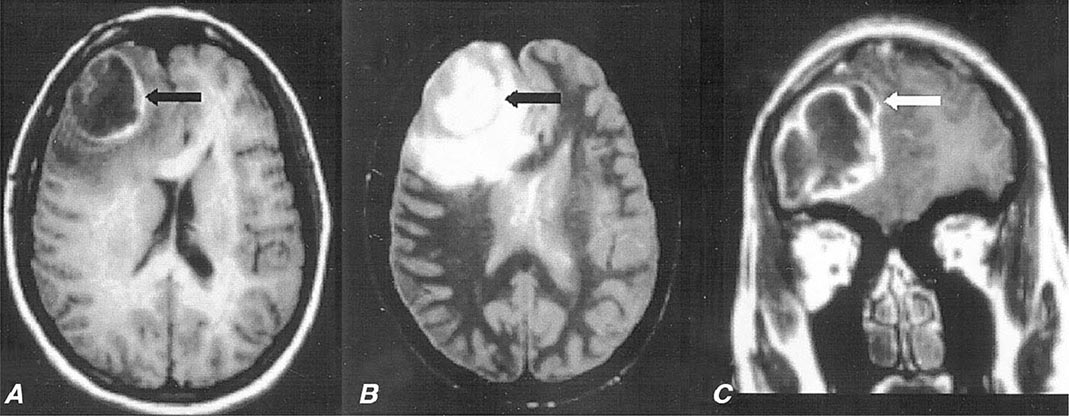
FIGURE 164-4 Pneumococcal brain abscess. Note that the abscess wall has hyperintense signal on the axial T1-weighted magnetic resonance imaging (MRI) (A, black arrow), hypointense signal on the axial proton density images (B, black arrow), and enhances prominently after gadolinium administration on the coronal T1-weighted image (C). The abscess is surrounded by a large amount of vasogenic edema and has a small “daughter” abscess (C, white arrow). (Courtesy of Joseph Lurito, MD; with permission.)
Microbiologic diagnosis of the etiologic agent is most accurately determined by Gram’s stain and culture of abscess material obtained by CT-guided stereotactic needle aspiration. Aerobic and anaerobic bacterial cultures and mycobacterial and fungal cultures should be obtained. Up to 10% of patients will also have positive blood cultures. LP should not be performed in patients with known or suspected focal intracranial infections such as abscess or empyema; CSF analysis contributes nothing to diagnosis or therapy, and LP increases the risk of herniation.
Additional laboratory studies may provide clues to the diagnosis of brain abscess in patients with a CNS mass lesion. About 50% of patients have a peripheral leukocytosis, 60% an elevated ESR, and 80% an elevated C-reactive protein. Blood cultures are positive in ~10% of cases overall but may be positive in >85% of patients with abscesses due to Listeria.
DIFFERENTIAL DIAGNOSIS
Conditions that can cause headache, fever, focal neurologic signs, and seizure activity include brain abscess, subdural empyema, bacterial meningitis, viral meningoencephalitis, superior sagittal sinus thrombosis, and acute disseminated encephalomyelitis. When fever is absent, primary and metastatic brain tumors become the major differential diagnosis. Less commonly, cerebral infarction or hematoma can have an MRI or CT appearance resembling brain abscess.
TREATMENT | BRAIN ABSCESS |
Optimal therapy of brain abscesses involves a combination of high-dose parenteral antibiotics and neurosurgical drainage. Empirical therapy of community-acquired brain abscess in an immunocompetent patient typically includes a third- or fourth-generation cephalosporin (e.g., cefotaxime, ceftriaxone, or cefepime) and metronidazole (see Table 164-1 for antibiotic dosages). In patients with penetrating head trauma or recent neurosurgical procedures, treatment should include ceftazidime as the third-generation cephalosporin to enhance coverage of Pseudomonas spp. and vancomycin for coverage of staphylococci. Meropenem plus vancomycin also provides good coverage in this setting.
Aspiration and drainage of the abscess under stereotactic guidance are beneficial for both diagnosis and therapy. Empirical antibiotic coverage should be modified based on the results of Gram’s stain and culture of the abscess contents. Complete excision of a bacterial abscess via craniotomy or craniectomy is generally reserved for multiloculated abscesses or those in which stereotactic aspiration is unsuccessful.
Medical therapy alone is not optimal for treatment of brain abscess and should be reserved for patients whose abscesses are neurosurgically inaccessible, for patients with small (<2–3 cm) or nonencapsulated abscesses (cerebritis), and for patients whose condition is too tenuous to allow performance of a neurosurgical procedure. All patients should receive a minimum of 6–8 weeks of parenteral antibiotic therapy. The role, if any, of supplemental oral antibiotic therapy following completion of a standard course of parenteral therapy has never been adequately studied.
In addition to surgical drainage and antibiotic therapy, patients should receive prophylactic anticonvulsant therapy because of the high risk (~35%) of focal or generalized seizures. Anticonvulsant therapy is continued for at least 3 months after resolution of the abscess, and decisions regarding withdrawal are then based on the EEG. If the EEG is abnormal, anticonvulsant therapy should be continued. If the EEG is normal, anticonvulsant therapy can be slowly withdrawn, with close follow-up and repeat EEG after the medication has been discontinued.
Glucocorticoids should not be given routinely to patients with brain abscesses. Intravenous dexamethasone therapy (10 mg every 6 h) is usually reserved for patients with substantial periabscess edema and associated mass effect and increased ICP. Dexamethasone should be tapered as rapidly as possible to avoid delaying the natural process of encapsulation of the abscess.
Serial MRI or CT scans should be obtained on a monthly or twice-monthly basis to document resolution of the abscess. More frequent studies (e.g., weekly) are probably warranted in the subset of patients who are receiving antibiotic therapy alone. A small amount of enhancement may remain for months after the abscess has been successfully treated.
PROGNOSIS
The mortality rate of brain abscess has declined in parallel with the development of enhanced neuroimaging techniques, improved neurosurgical procedures for stereotactic aspiration, and improved antibiotics. In modern series, the mortality rate is typically <15%. Significant sequelae, including seizures, persisting weakness, aphasia, or mental impairment, occur in ≥20% of survivors.
NONBACTERIAL CAUSES OF INFECTIOUS FOCAL CNS LESIONS
ETIOLOGY
Neurocysticercosis is the most common parasitic disease of the CNS worldwide. Humans acquire cysticercosis by the ingestion of food contaminated with the eggs of the parasite T. solium. Toxoplasmosis is a parasitic disease caused by T. gondii and acquired from the ingestion of undercooked meat and from handling cat feces.
CLINICAL PRESENTATION
The most common manifestation of neurocysticercosis is new-onset partial seizures with or without secondary generalization. Cysticerci may develop in the brain parenchyma and cause seizures or focal neurologic deficits. When present in the subarachnoid or ventricular spaces, cysticerci can produce increased ICP by interference with CSF flow. Spinal cysticerci can mimic the presentation of intraspinal tumors. When the cysticerci first lodge in the brain, they frequently cause little in the way of an inflammatory response. As the cysticercal cyst degenerates, it elicits an inflammatory response that may present clinically as a seizure. Eventually the cyst dies, a process that may take several years and is typically associated with resolution of the inflammatory response and, often, abatement of seizures.
Primary Toxoplasma infection is often asymptomatic. However, during this phase parasites may spread to the CNS, where they become latent. Reactivation of CNS infection is almost exclusively associated with immunocompromised hosts, particularly those with HIV infection. During this phase patients present with headache, fever, seizures, and focal neurologic deficits.
DIAGNOSIS
The lesions of neurocysticercosis are readily visualized by MRI or CT scans. Lesions with viable parasites appear as cystic lesions. The scolex can often be visualized on MRI. Lesions may appear as contrast-enhancing lesions surrounded by edema. A very early sign of cyst death is hypointensity of the vesicular fluid on T2-weighted images when compared with CSF. Parenchymal brain calcifications are the most common finding and evidence that the parasite is no longer viable. MRI findings of toxoplasmosis consist of multiple lesions in the deep white matter, the thalamus, and basal ganglia and at the gray-white junction in the cerebral hemispheres. With contrast administration, the majority of the lesions enhance in a ringed, nodular, or homogeneous pattern and are surrounded by edema. In the presence of the characteristic neuroimaging abnormalities of T. gondii infection, serum IgG antibody to T. gondii should be obtained and, when positive, the patient should be treated.
TREATMENT | INFECTIOUS FOCAL CNS LESIONS |
Anticonvulsant therapy is initiated when the patient with neurocysticercosis presents with a seizure. There is controversy about whether or not anthelmintic therapy should be given to all patients, and recommendations are based on the stage of the lesion. Cysticerci appearing as cystic lesions in the brain parenchyma with or without pericystic edema or in the subarachnoid space at the convexity of the cerebral hemispheres should be treated with anticysticidal therapy. Cysticidal drugs accelerate the destruction of the parasites, resulting in a faster resolution of the infection. Albendazole and praziquantel are used in the treatment of neurocysticercosis. Approximately 85% of parenchymal cysts are destroyed by a single course of albendazole, and ~75% are destroyed by a single course of praziquantel. The dose of albendazole is 15 mg/kg per day in two doses for 8 days. The dose of praziquantel is 50 mg/kg per day for 15 days, although a number of other dosage regimens are also frequently cited. Prednisone or dexamethasone is given with anticysticidal therapy to reduce the host inflammatory response to degenerating parasites. Many, but not all, experts recommend anticysticidal therapy for lesions that are surrounded by a contrast-enhancing ring. There is universal agreement that calcified lesions do not need to be treated with anticysticidal therapy. Antiepileptic therapy can be stopped once the follow-up CT scan shows resolution of the lesion. Long-term antiepileptic therapy is recommended when seizures occur after resolution of edema and resorption or calcification of the degenerating cyst.
CNS toxoplasmosis is treated with a combination of sulfadiazine, 1.5–2.0 g orally qid, plus pyrimethamine, 100 mg orally to load, then 75–100 mg orally qd, plus folinic acid, 10–15 mg orally qd. Folinic acid is added to the regimen to prevent megaloblastic anemia. Therapy is continued until there is no evidence of active disease on neuroimaging studies, which typically takes at least 6 weeks, and then the dose of sulfadiazine is reduced to 2–4 g/d and pyrimethamine to 50 mg/d. Clindamycin plus pyrimethamine is an alternative therapy for patients who cannot tolerate sulfadiazine, but the combination of pyrimethamine and sulfadiazine is more effective.
SUBDURAL EMPYEMA
A subdural empyema (SDE) is a collection of pus between the dura and arachnoid membranes (Fig. 164-5).
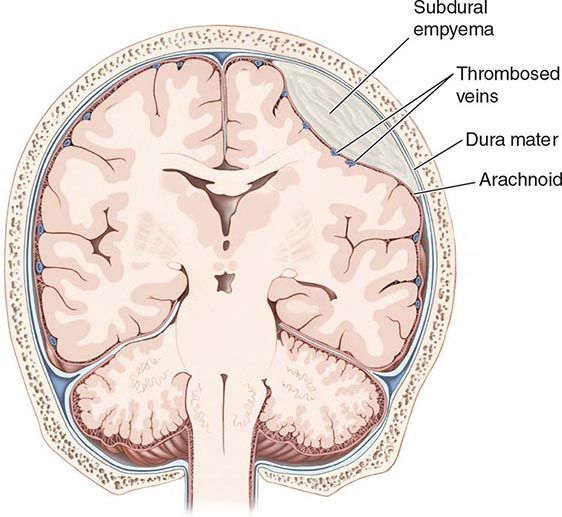
FIGURE 164-5 Subdural empyema.
EPIDEMIOLOGY
SDE is a rare disorder that accounts for 15–25% of focal suppurative CNS infections. Sinusitis is the most common predisposing condition and typically involves the frontal sinuses, either alone or in combination with the ethmoid and maxillary sinuses. Sinusitis-associated empyema has a striking predilection for young males, possibly reflecting sex-related differences in sinus anatomy and development. It has been suggested that SDE may complicate 1–2% of cases of frontal sinusitis severe enough to require hospitalization. As a consequence of this epidemiology, SDE shows an ~3:1 male/female predominance, with 70% of cases occurring in the second and third decades of life. SDE may also develop as a complication of head trauma or neurosurgery. Secondary infection of a subdural effusion may also result in empyema, although secondary infection of hematomas, in the absence of a prior neurosurgical procedure, is rare.
ETIOLOGY
Aerobic and anaerobic streptococci, staphylococci, Enterobacteriaceae, and anaerobic bacteria are the most common causative organisms of sinusitis-associated SDE. Staphylococci and gram-negative bacilli are often the etiologic organisms when SDE follows neurosurgical procedures or head trauma. Up to one-third of cases are culture-negative, possibly reflecting difficulty in obtaining adequate anaerobic cultures.
PATHOPHYSIOLOGY
Sinusitis-associated SDE develops as a result of either retrograde spread of infection from septic thrombophlebitis of the mucosal veins draining the sinuses or contiguous spread of infection to the brain from osteomyelitis in the posterior wall of the frontal or other sinuses. SDE may also develop from direct introduction of bacteria into the subdural space as a complication of a neurosurgical procedure. The evolution of SDE can be extremely rapid because the subdural space is a large compartment that offers few mechanical barriers to the spread of infection. In patients with sinusitis-associated SDE, suppuration typically begins in the upper and anterior portions of one cerebral hemisphere and then extends posteriorly. SDE is often associated with other intracranial infections, including epidural empyema (40%), cortical thrombophlebitis (35%), and intracranial abscess or cerebritis (>25%). Cortical venous infarction produces necrosis of underlying cerebral cortex and subcortical white matter, with focal neurologic deficits and seizures (see below).
CLINICAL PRESENTATION
A patient with SDE typically presents with fever and a progressively worsening headache. The diagnosis of SDE should always be suspected in a patient with known sinusitis who presents with new CNS signs or symptoms. Patients with underlying sinusitis frequently have symptoms related to this infection. As the infection progresses, focal neurologic deficits, seizures, nuchal rigidity, and signs of increased ICP commonly occur. Headache is the most common complaint at the time of presentation; initially it is localized to the side of the subdural infection, but then it becomes more severe and generalized. Contralateral hemiparesis or hemiplegia is the most common focal neurologic deficit and can occur from the direct effects of the SDE on the cortex or as a consequence of venous infarction. Seizures begin as partial motor seizures that then become secondarily generalized. Seizures may be due to the direct irritative effect of the SDE on the underlying cortex or result from cortical venous infarction (see above). In untreated SDE, the increasing mass effect and increase in ICP cause progressive deterioration in consciousness, leading ultimately to coma.
DIAGNOSIS
MRI (Fig. 164-6) is superior to CT in identifying SDE and any associated intracranial infections. The administration of gadolinium greatly improves diagnosis by enhancing the rim of the empyema and allowing the empyema to be clearly delineated from the underlying brain parenchyma. Cranial MRI is also extremely valuable in identifying sinusitis, other focal CNS infections, cortical venous infarction, cerebral edema, and cerebritis. CT may show a crescent-shaped hypodense lesion over one or both hemispheres or in the interhemispheric fissure. Frequently the degree of mass effect, exemplified by midline shift, ventricular compression, and sulcal effacement, is far out of proportion to the mass of the SDE.
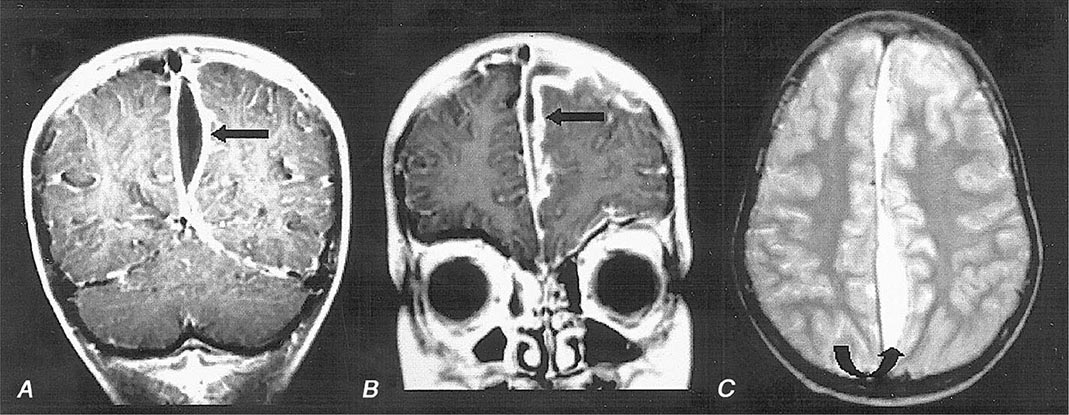
FIGURE 164-6 Subdural empyema. There is marked enhancement of the dura and leptomeninges (A, B, straight arrows) along the left medial hemisphere. The pus is hypointense on T1-weighted images (A, B) but markedly hyperintense on the proton density–weighted (C, curved arrow) image. (Courtesy of Joseph Lurito, MD; with permission.)
CSF examination should be avoided in patients with known or suspected SDE because it adds no useful information and is associated with the risk of cerebral herniation.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of the combination of headache, fever, focal neurologic signs, and seizure activity that progresses rapidly to an altered level of consciousness includes subdural hematoma, bacterial meningitis, viral encephalitis, brain abscess, superior sagittal sinus thrombosis, and acute disseminated encephalomyelitis. The presence of nuchal rigidity is unusual with brain abscess or epidural empyema and should suggest the possibility of SDE when associated with significant focal neurologic signs and fever. Patients with bacterial meningitis also have nuchal rigidity but do not typically have focal deficits of the severity seen with SDE.
TREATMENT | SUBDURAL EMPYEMA |
SDE is a medical emergency. Emergent neurosurgical evacuation of the empyema, either through craniotomy, craniectomy, or burr-hole drainage, is the definitive step in the management of this infection. Empirical antimicrobial therapy for community-acquired SDE should include a combination of a third-generation cephalosporin (e.g., cefotaxime or ceftriaxone), vancomycin, and metronidazole (see Table 164-1 for dosages). Patients with hospital-acquired SDE may have infections due to Pseudomonas spp. or MRSA and should receive coverage with a carbapenem (e.g., meropenem) and vancomycin. Metronidazole is not necessary for antianaerobic therapy when meropenem is being used. Parenteral antibiotic therapy should be continued for a minimum of 3–4 weeks after SDE drainage. Patients with associated cranial osteomyelitis may require longer therapy. Specific diagnosis of the etiologic organisms is made based on Gram’s stain and culture of fluid obtained via either burr holes or craniotomy; the initial empirical antibiotic coverage can be modified accordingly.
PROGNOSIS
Prognosis is influenced by the level of consciousness of the patient at the time of hospital presentation, the size of the empyema, and the speed with which therapy is instituted. Long-term neurologic sequelae, which include seizures and hemiparesis, occur in up to 50% of cases.
CRANIAL EPIDURAL ABSCESS
Cranial epidural abscess is a suppurative infection occurring in the potential space between the inner skull table and dura (Fig. 164-7).
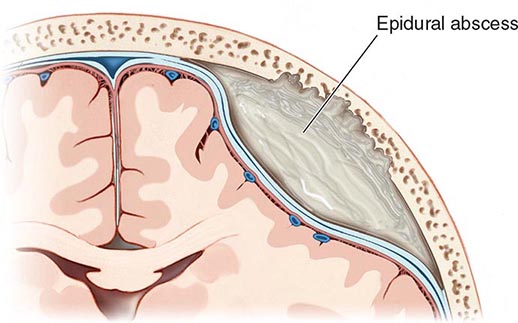
FIGURE 164-7 Cranial epidural abscess is a collection of pus between the dura and the inner table of the skull.
ETIOLOGY AND PATHOPHYSIOLOGY
Cranial epidural abscess is less common than either brain abscess or SDE and accounts for <2% of focal suppurative CNS infections. A cranial epidural abscess develops as a complication of a craniotomy or compound skull fracture or as a result of spread of infection from the frontal sinuses, middle ear, mastoid, or orbit. An epidural abscess may develop contiguous to an area of osteomyelitis, when craniotomy is complicated by infection of the wound or bone flap, or as a result of direct infection of the epidural space. Infection in the frontal sinus, middle ear, mastoid, or orbit can reach the epidural space through retrograde spread of infection from septic thrombophlebitis in the emissary veins that drain these areas or by way of direct spread of infection through areas of osteomyelitis. Unlike the subdural space, the epidural space is really a potential rather than an actual compartment. The dura is normally tightly adherent to the inner skull table, and infection must dissect the dura away from the skull table as it spreads. As a result, epidural abscesses are often smaller than SDEs. Cranial epidural abscesses, unlike brain abscesses, only rarely result from hematogenous spread of infection from extracranial primary sites. The bacteriology of a cranial epidural abscess is similar to that of SDE (see above). The etiologic organisms of an epidural abscess that arises from frontal sinusitis, middle-ear infections, or mastoiditis are usually streptococci or anaerobic organisms. Staphylococci or gram-negative organisms are the usual cause of an epidural abscess that develops as a complication of craniotomy or compound skull fracture.
CLINICAL PRESENTATION
Patients present with fever (60%), headache (40%), nuchal rigidity (35%), seizures (10%), and focal deficits (5%). Development of symptoms may be insidious, as the empyema usually enlarges slowly in the confined anatomic space between the dura and the inner table of the skull. Periorbital edema and Pott’s puffy tumor, reflecting underlying associated frontal bone osteomyelitis, are present in ~40%. In patients with a recent neurosurgical procedure, wound infection is invariably present, but other symptoms may be subtle and can include altered mental status (45%), fever (35%), and headache (20%). The diagnosis should be considered when fever and headache follow recent head trauma or occur in the setting of frontal sinusitis, mastoiditis, or otitis media.
DIAGNOSIS
Cranial MRI with gadolinium enhancement is the procedure of choice to demonstrate a cranial epidural abscess. The sensitivity of CT is limited by the presence of signal artifacts arising from the bone of the inner skull table. The CT appearance of an epidural empyema is that of a lens or crescent-shaped hypodense extraaxial lesion. On MRI, an epidural empyema appears as a lentiform or crescent-shaped fluid collection that is hyperintense compared to CSF on T2-weighted images. On T1-weighted images, the fluid collection may be either isointense or hypointense compared to brain. Following the administration of gadolinium, there is linear enhancement of the dura on T1-weighted images. In distinction to subdural empyema, signs of mass effect or other parenchymal abnormalities are uncommon.
TREATMENT | EPIDURAL ABSCESS |
Immediate neurosurgical drainage is indicated. Empirical antimicrobial therapy, pending the results of Gram’s stain and culture of the purulent material obtained at surgery, should include a combination of a third-generation cephalosporin, vancomycin, and metronidazole (Table 164-1). Ceftazidime or meropenem should be substituted for ceftriaxone or cefotaxime in neurosurgical patients. Metronidazole is not necessary for antianaerobic coverage in patients receiving meropenem. When the organism has been identified, antimicrobial therapy can be modified accordingly. Antibiotics should be continued for 3–6 weeks after surgical drainage. Patients with associated osteomyelitis may require additional therapy.
PROGNOSIS
The mortality rate is <5% in modern series, and full recovery is the rule in most survivors.
SUPPURATIVE THROMBOPHLEBITIS
DEFINITION
Suppurative intracranial thrombophlebitis is septic venous thrombosis of cortical veins and sinuses. This may occur as a complication of bacterial meningitis; SDE; epidural abscess; or infection in the skin of the face, paranasal sinuses, middle ear, or mastoid.
ANATOMY AND PATHOPHYSIOLOGY
The cerebral veins and venous sinuses have no valves; therefore, blood within them can flow in either direction. The superior sagittal sinus is the largest of the venous sinuses (Fig. 164-8). It receives blood from the frontal, parietal, and occipital superior cerebral veins and the diploic veins, which communicate with the meningeal veins. Bacterial meningitis is a common predisposing condition for septic thrombosis of the superior sagittal sinus. The diploic veins, which drain into the superior sagittal sinus, provide a route for the spread of infection from the meninges, especially in cases where there is purulent exudate near areas of the superior sagittal sinus. Infection can also spread to the superior sagittal sinus from nearby SDE or epidural abscess. Dehydration from vomiting, hypercoagulable states, and immunologic abnormalities, including the presence of circulating antiphospholipid antibodies, also contribute to cerebral venous sinus thrombosis. Thrombosis may extend from one sinus to another, and at autopsy, thrombi of different histologic ages can often be detected in several sinuses. Thrombosis of the superior sagittal sinus is often associated with thrombosis of superior cortical veins and small parenchymal hemorrhages.
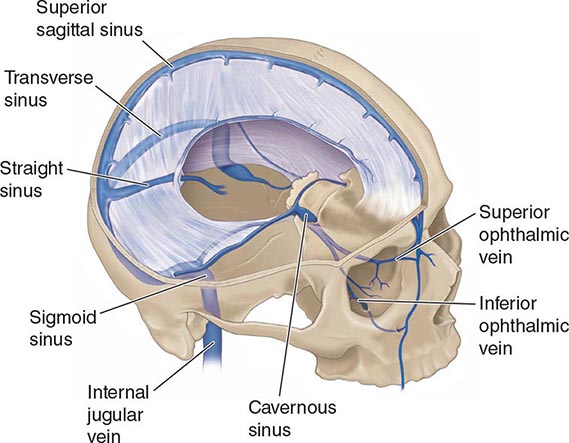
FIGURE 164-8 Anatomy of the cerebral venous sinuses.
The superior sagittal sinus drains into the transverse sinuses (Fig. 164-8). The transverse sinuses also receive venous drainage from small veins from both the middle ear and mastoid cells. The transverse sinus becomes the sigmoid sinus before draining into the internal jugular vein. Septic transverse/sigmoid sinus thrombosis can be a complication of acute and chronic otitis media or mastoiditis. Infection spreads from the mastoid air cells to the transverse sinus via the emissary veins or by direct invasion. The cavernous sinuses are inferior to the superior sagittal sinus at the base of the skull. The cavernous sinuses receive blood from the facial veins via the superior and inferior ophthalmic veins. Bacteria in the facial veins enter the cavernous sinus via these veins. Bacteria in the sphenoid and ethmoid sinuses can spread to the cavernous sinuses via the small emissary veins. The sphenoid and ethmoid sinuses are the most common sites of primary infection resulting in septic cavernous sinus thrombosis.
CLINICAL MANIFESTATIONS
Septic thrombosis of the superior sagittal sinus presents with headache, fever, nausea and vomiting, confusion, and focal or generalized seizures. There may be a rapid development of stupor and coma. Weakness of the lower extremities with bilateral Babinski’s signs or hemiparesis is often present. When superior sagittal sinus thrombosis occurs as a complication of bacterial meningitis, nuchal rigidity and Kernig’s and Brudzinski’s signs may be present.
The oculomotor nerve, the trochlear nerve, the abducens nerve, the ophthalmic and maxillary branches of the trigeminal nerve, and the internal carotid artery all pass through the cavernous sinus (see Fig. 455-4). The symptoms of septic cavernous sinus thrombosis are fever, headache, frontal and retroorbital pain, and diplopia. The classic signs are ptosis, proptosis, chemosis, and extraocular dysmotility due to deficits of cranial nerves III, IV, and VI; hyperesthesia of the ophthalmic and maxillary divisions of the fifth cranial nerve and a decreased corneal reflex may be detected. There may be evidence of dilated, tortuous retinal veins and papilledema.
Headache and earache are the most frequent symptoms of transverse sinus thrombosis. A transverse sinus thrombosis may also present with otitis media, sixth nerve palsy, and retroorbital or facial pain (Gradenigo’s syndrome). Sigmoid sinus and internal jugular vein thrombosis may present with neck pain.
DIAGNOSIS
The diagnosis of septic venous sinus thrombosis is suggested by an absent flow void within the affected venous sinus on MRI and confirmed by magnetic resonance venography, CT angiogram, or the venous phase of cerebral angiography. The diagnosis of thrombophlebitis of intracerebral and meningeal veins is suggested by the presence of intracerebral hemorrhage but requires cerebral angiography for definitive diagnosis.
TREATMENT | SUPPURATIVE THROMBOPHLEBITIS |
Septic venous sinus thrombosis is treated with antibiotics, hydration, and removal of infected tissue and thrombus in septic lateral or cavernous sinus thrombosis. The choice of antimicrobial therapy is based on the bacteria responsible for the predisposing or associated condition. Optimal duration of therapy is unknown, but antibiotics are usually continued for 6 weeks or until there is radiographic evidence of resolution of thrombosis. Anticoagulation with dose-adjusted intravenous heparin is recommended for aseptic venous sinus thrombosis and in the treatment of septic venous sinus thrombosis complicating bacterial meningitis in patients who have progressive neurologic deterioration despite antimicrobial therapy and intravenous fluids. The presence of a small intracerebral hemorrhage from septic thrombophlebitis is not an absolute contraindication to heparin therapy. Successful management of aseptic venous sinus thrombosis has been reported with surgical thrombectomy, catheter-directed urokinase therapy, and a combination of intrathrombus recombinant tissue plasminogen activator (rtPA) and intravenous heparin, but there are not enough data to recommend these therapies in septic venous sinus thrombosis.
165 | Chronic and Recurrent Meningitis |
Chronic inflammation of the meninges (pia, arachnoid, and dura) can produce profound neurologic disability and may be fatal if not successfully treated. Chronic meningitis is diagnosed when a characteristic neurologic syndrome exists for >4 weeks and is associated with a persistent inflammatory response in the cerebrospinal fluid (CSF) (white blood cell count >5/μL). The causes are varied, and appropriate treatment depends on identification of the etiology. Five categories of disease account for most cases of chronic meningitis: (1) meningeal infections, (2) malignancy, (3) autoimmune inflammatory disorders, (4) chemical meningitis, and (5) parameningeal infections.
CLINICAL PATHOPHYSIOLOGY
Neurologic manifestations of chronic meningitis (Table 165-1) are determined by the anatomic location of the inflammation and its consequences. Persistent headache with or without a stiff neck, hydrocephalus, cranial neuropathies, radiculopathies, and cognitive or personality changes are the cardinal features. These can occur alone or in combination. When they appear in combination, widespread dissemination of the inflammatory process along CSF pathways has occurred. In some cases, the presence of an underlying systemic illness points to a specific agent or class of agents as the probable cause. The diagnosis of chronic meningitis is usually made when the clinical presentation prompts the physician to examine the CSF for signs of inflammation. CSF is produced by the choroid plexus of the cerebral ventricles, exits through narrow foramina into the subarachnoid space surrounding the brain and spinal cord, circulates around the base of the brain and over the cerebral hemispheres, and is resorbed by arachnoid villi projecting into the superior sagittal sinus. CSF flow provides a pathway for rapid spread of infectious and other infiltrative processes over the brain, spinal cord, and cranial and spinal nerve roots. Spread from the subarachnoid space into brain parenchyma may occur via the arachnoid cuffs that surround blood vessels that penetrate brain tissue (Virchow-Robin spaces).
SYMPTOMS AND SIGNS OF CHRONIC MENINGITIS |
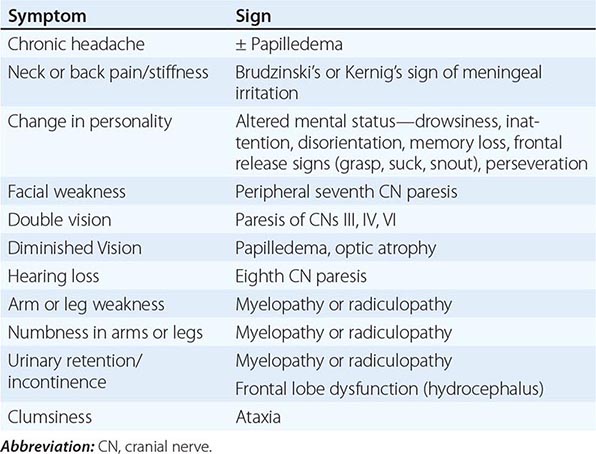
Intracranial Meningitis Nociceptive nerve fibers of the meninges are stimulated by the inflammatory process, resulting in headache, neck pain, or back pain. Obstruction of CSF pathways at the foramina or arachnoid villi may produce hydrocephalus and symptoms of raised intracranial pressure (ICP), including headache, vomiting, apathy or drowsiness, gait instability, papilledema, visual loss, impaired upgaze, or palsy of the sixth cranial nerve (CN) (Chap. 455). Cognitive and behavioral changes during the course of chronic meningitis may also result from vascular damage, which may similarly produce seizures, stroke, or myelopathy. Inflammatory deposits seeded via the CSF circulation are often prominent around the brainstem and cranial nerves and along the undersurface of the frontal and temporal lobes. Such cases, termed basal meningitis, often present as multiple cranial neuropathies, with decreased vision (CN II), facial weakness (CN VII), decreased hearing (CN VIII), diplopia (CNs III, IV, and VI), sensory or motor abnormalities of the oropharynx (CNs IX, X, and XII), decreased olfaction (CN I), or decreased facial sensation and masseter weakness (CN V).
Spinal Meningitis Injury may occur to motor and sensory roots as they traverse the subarachnoid space and penetrate the meninges. These cases present as multiple radiculopathies with combinations of radicular pain, sensory loss, motor weakness, and urinary or fecal incontinence. Meningeal inflammation can encircle the cord, resulting in a myelopathy. Patients with slowly progressive involvement of multiple cranial nerves and/or spinal nerve roots are likely to have chronic meningitis. Electrophysiologic testing (electromyography, nerve conduction studies, and evoked response testing) may be helpful in determining whether there is involvement of cranial and spinal nerve roots.
Systemic Manifestations In some patients, evidence of systemic disease provides clues to the underlying cause of chronic meningitis. A complete history of travel, sexual practice, and exposure to infectious agents should be sought. Infectious causes are often associated with fever, malaise, anorexia, and signs of localized or disseminated infection outside the nervous system. Infectious causes are of major concern in the immunosuppressed patient, especially in patients with AIDS, in whom chronic meningitis may present without headache or fever. Noninfectious inflammatory disorders often produce systemic manifestations, but meningitis may be the initial manifestation. Carcinomatous meningitis may or may not be accompanied by clinical evidence of the primary neoplasm.



