morphologic alterations have no apparent clinical consequence. In other disorders—such as tyrosinemia and glycogen storage disease (GSD) IV—progressive liver disease is common. A variety of histologic alterations—including hepatitis, steatosis, cirrhosis, cholestasis, ductopenia, ductular proliferation, neoplasia, and storage—can be seen in IEM, and many IEM cause similar morphologic alterations (see Chapter 15).
TABLE 5-1 CLINICAL SYMPTOMS IN PATIENTS WITH INBORN ERRORS OF METABOLISM | |
|---|---|
|
TABLE 5-2 PLACENTAL FINDINGS IN LYSOSOMAL STORAGE DISEASES | |||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
that every NBS program includes a uniform screening panel (Table 5-4) and advises on the addition of new tests to the screening panel (http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/). The Web site has a list of conditions that have been nominated and are under current review.
TABLE 5-3 ULTRASTRUCTURAL APPEARANCE OF LYSOSOMAL STORAGE DISEASES IN SKIN AND CONJUNCTIVAL BIOPSIES | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
includes the use of molecular techniques to detect a panel of common mutations. The initial screen is done by measurement of immunoreactive trypsinogen and then followed by molecular genetic testing of the CFTR gene and sweat testing. The genetic testing involves determination of a panel of common mutations, and a diagnosis is made based on a combination of sweat test results and genetic testing (5).
TABLE 5-4 RECOMMENDED UNIFORM SCREENING PANEL FOR ALL STATES | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
TABLE 5-5 MASON CATEGORIES OF LYSOSOMAL STORAGE DISORDERS | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
The central and peripheral nervous systems are particularly vulnerable to lysosomal storage diseases and therefore often affected. Neurologic manifestations include developmental delay, dementia, speech impairment, deafness, visual loss, ophthalmoplegia, leukodystrophy, seizures, ataxia, peripheral neuropathy, and macrocephaly (15).
Hepatosplenomegaly is a feature of many of these diseases including Gaucher disease, Niemann-Pick disease, and mucopolysaccharidosis (MPS). To what extent Kupffer cells or hepatocytes are affected varies depending on the disease process.
Deposition of storage material like glycosaminoglycans in growth plates and cartilage disrupts normal development and maturation of the skeleton. As a result, these patients often have coarse, dysmorphic facial features. “Dysostosis multiplex” is a term that is often used for the skeletal manifestations that include coarse, dysmorphic facial features, short stature, as well as bone and joint abnormalities.
Toxic effect: At least in some cases, cellular and neuronal dysfunction is independent of neuronal storage. In Krabbe disease, secondary increase of the lysosphingolipid psychosine is thought to have toxic effects (12).
Vesicle function and trafficking: Lysosomes are connected to many key vesicle and cell trafficking systems, including autophagy and exocytosis. Lysosomal storage diseases lead to disruption of pH gradients in the endomembrane system that can disrupt function of many proteins (18). Autophagy and exocytosis may be disrupted (18).
Lipid composition: Changes in the composition of lipids in cell membranes can alter membrane fluidity and affect receptor signaling. This is, for example, observed in Niemann-Pick disease type C.
Signaling and activation of apoptosis: Some of the accumulating storage products can act as ligands for cellular receptors. Examples include galactoceramide in Krabbe and glycosaminoglycans in mucopolysaccharidoses. These disruptions in cell signaling can lead to activation of proapoptotic pathways (12). Activation of the unfolded protein response resulting from disrupted function of the endoplasmic reticulum can also be a pathway leading to apoptosis.
Inflammation: Niemann-Pick disease (types A, B, C) and Gaucher disease as diseases associated with prominent involvement of the reticuloendothelial system can lead to release of inflammatory cytokines from cells like microglial cells.
Calcium homeostasis: Storage products can disrupt calcium handling in the endoplasmic reticulum affecting cellular calcium homeostasis.
Enzyme replacement therapies are approved and used for Pompe disease, adult-onset acid maltase deficiency, non-neuropathic type I Gaucher disease, Fabry disease, MPS I S, MPS I S/H, MPS II, and MPS VI. Replacement therapies for alpha-mannosidosis, GM1 gangliosidosis, Wolman disease, and MPS IV A are in clinical trials. Studies with intrathecal administration and with combination therapies are ongoing (12,18,27,28).
Substrate reduction therapies aim to reduce the upstream synthesis of accumulating storage products. Miglustat (N-butyl-deoxygalactonojirimycin) is a competitive inhibitor of ceramide glucosyltransferase and is of potential use in the treatment of Gaucher I and III as well as Niemann-Pick disease type C (27).
Pharmacologic chaperones stabilize a protein during the transport and processing in the endoplasmic reticulum, preventing mutant protein from being sent for early degradation and allowing for the expression of mutant proteins with residual activity. Miglustat has shown beneficial effects in mouse models of Fabry disease (27).
Bone marrow transplantation has been shown to benefit patients with Krabbe disease and Hurler disease and some patients with late-onset metachromatic leukodystrophy if performed before onset of symptoms. Skeletal dysplasia in Hurler disease is, however, not fully corrected, and treated children with Krabbe disease may still develop manifestations such as motor difficulty (15,29). Gene therapy is also being studied as another approach to treatment (30).
cardiac disease, or cerebrovascular disease (38). Late-onset manifestations with cardiac disease with or without renal involvement can be seen even into the 6th decade, and the above cited incidence may underestimate the overall disease burden (37,39). In contrast to other X-linked diseases, the phenotype in female heterozygotes is variable, but can be associated with significant medical problems mimicking those of the classic disease seen in males (40). Hemizygotes and heterozygotes with B or AB blood type are more severely affected due to the additional accumulation of B-specific glycolipid.
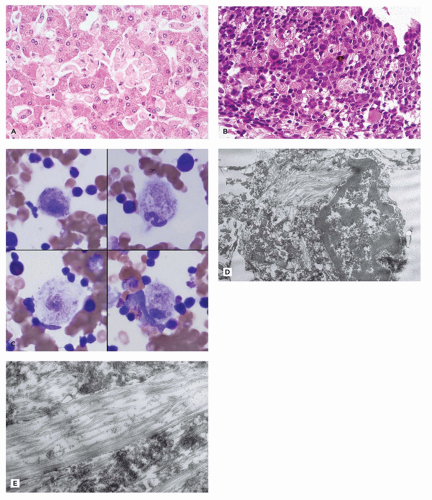 FIGURE 5-1 • Gaucher disease. A: The liver of a patient with Gaucher disease has prominent Kupffer cells due to pale eosinophilic expansion of the cytoplasm by lysosomal glucocerebroside storage material (H&E). B: Enlarged phagocytes in the spleen have a “wrinkled tissue paper” appearance of their cytoplasm because of the glucocerebroside storage (H&E). C: Wright-stained bone marrow aspirate from a patient with Gaucher disease showing “Gaucher cells” with a “wrinkled tissue paper” appearance of cytoplasm due to glucocerebroside storage (Wright). D: Ultrastructural appearance of Gaucher cell from the spleen, obtained at autopsy, showing cytoplasmic storage in upper middle and lower middle of the image, to the left of the nucleus (Uranyl acetate, lead citrate). E: Ultrastructurally, glucocerebroside storage material in Gaucher disease comprises rod-shaped or tubular lipid bilayer stacks with a diameter of up to 4 µm (Uranyl acetate, lead citrate). |
and tubules are seen in endothelial, perithelial, and smooth muscle cells (41,42). Glomerular podocytes, endothelial, mesangial, interstitial, and tubular epithelial cells all contain storage material. Podocyte storage causes cellular injury, followed by glomerular capillary wall thickening, progressive mesangial matrix expansion, glomerulosclerosis, and, eventually, end-stage renal disease (Figure 5-2A to C). Some patients develop hypertrophic obstructive cardiomyopathy. Storage material in arteries can lead to luminal obstruction and ischemic lesions that may, for example, be found in the brain. There is typically no clinically significant liver involvement, but morphologic studies may show Kupffer cells that have a tan appearance in H&E sections and storage that is birefringent and crystalline in frozen sections stained with the Schultz method.
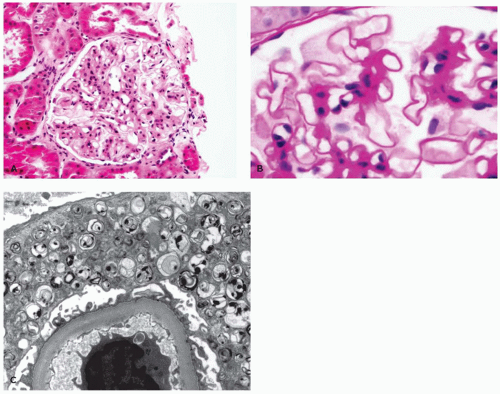 FIGURE 5-2 • Fabry disease. A: By LM, glomeruli in Fabry disease show mesangial expansion with prominence of pale-staining visceral epithelial cells (podocytes) (H&E). B: At higher magnification, podocyte cytoplasm is markedly expanded by PAS-positive storage material (PAS). C: Ultrastructurally, visceral epithelial cell cytoplasm is expanded by osmiophilic, lamellated leaflets and tubules, representing glycolipid and cholesterol storage (Uranyl acetate, lead citrate). (Images courtesy of Helen Liapis, M.D., Nephropath, Little Rock, AR.) |
endothelial cells, placental syncytiotrophoblasts, marrow histiocytes, lymphocytes, lymph nodes, thymus, lung, intestine, pancreas, pituitary, thyroid, salivary gland, skin (including sweat glands), and conjunctiva (45,46). Visceral storage material is fibrillogranular (45,46). Definitive diagnosis rests on demonstrating beta-galactosidase deficiency in leukocytes, fibroblasts, or amniocytes or on DNA analysis.
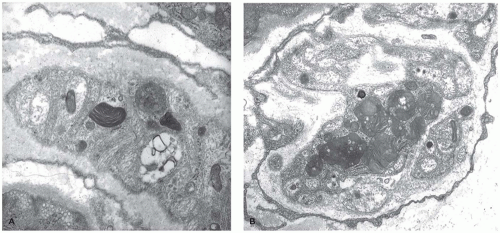 FIGURE 5-3 • Gangliosidosis. A, B: Membranous cytoplasmic bodies in GM2 (AB variant) gangliosidosis are heterogeneous and can show concentric or parallel structure, here in peripheral nerve axons. Though the morphology of the stored gangliosides is often not helpful in distinguishing the gangliosidoses, location of the storage material can be helpful (Uranyl acetate, lead citrate). (Image B from Vogler C, Rosenberg HS, Williams JC, et al. Electron microscopy in the diagnosis of lysosomal storage diseases. Am J Med Genet Suppl 1987;3:243-255, with permission.) |
aniline dyes that gave rise to the historical naming of this entity. An atypical form is caused by mutations in the cofactor saposin B (69). Different mutations have been described, and patients have a variable course with progressive neurologic disease resulting in several clinical forms with infantile, juvenile, and adult types. The central and peripheral nervous systems show demyelination, and the cerebellum is atrophic with Purkinje and granular cell loss. Spherical aggregates of metachromatic material that are 15 to 20 µm in diameter occur in oligodendrocytes, in macrophages within Virchow-Robin spaces, and in Schwann cells. This material comprises sulfatide, cholesterol, and phosphatides. In frozen sections, it stains positive with PAS, Alcian blue, and colloidal iron, and it is brown metachromatic (with 1% cresyl violet at low pH) and stains purple with toluidine blue. On electron microscopic studies, the storage material in oligodendrocytes, astrocytes, and Schwann cells is composed of closely packed, lamellar, amorphous, or prismatic material with alternating leaflets and tubules giving it a “herringbone” or “tuffstone” pattern (Figure 5-5A,B).
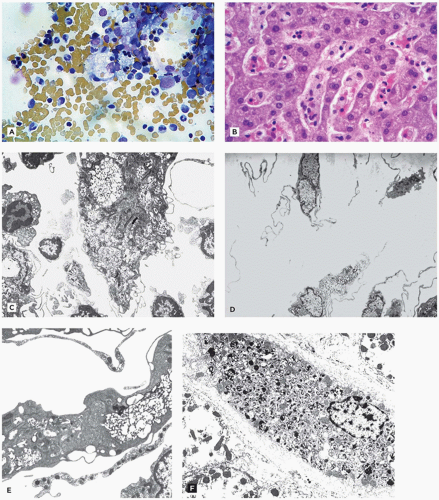 FIGURE 5-4 • Niemann-Pick disease. A: Several vacuolated “Niemann-Pick” cells, the pathologic hallmark of types A and B Niemann-Pick disease, are present and show “sea blue” coloration by Wright-Giemsa stain in this smear of a bone marrow aspirate. Niemann-Pick cells are capable of erythrophagocytosis and emperipolesis. B: Enlarged, foamy Kupffer cells in Niemann-Pick disease, as shown here, may be absent in very young children but become more prominent with time (H&E). C-F: Ultrastructurally, storage material in Niemann-Pick disease is a heterogeneous mix of membranous lamellar material, concentrically lamellated myelin-like material, and lipofuscin (C-F: Uranyl acetate, lead citrate). |
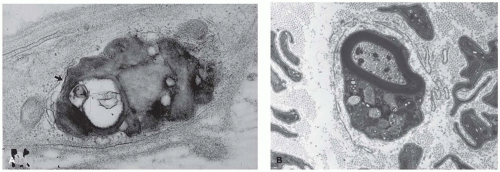 FIGURE 5-5 • Metachromatic leukodystrophy. A: This unmyelinated nerve from a conjunctival biopsy contains an inclusion of variable electron density. In some foci (arrow), closely approximated osmiophilic lamellae contribute to a subtle herringbone pattern. B: Myelinated nerve with pleomorphic lysosomes of variable density, “tuffstone” inclusions from a sural nerve of a patient with metachromatic leukodystrophy. (A, B: Uranyl acetate, lead citrate, A: Used from Vogler C, Rosenberg HS, Williams JC, et al. Electron microscopy in the diagnosis of lysosomal storage diseases. Am J Med Genet Suppl 1987;3:243-255, with permission.) |
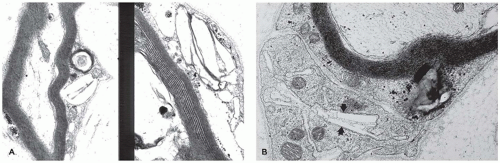 FIGURE 5-6 • Krabbe disease. A, B: Electron-lucent, angulated, and needle-shaped inclusions in conjunctival myelinated nerve Schwann cells, characteristic of Krabbe disease. (A, B: Uranyl acetate, lead citrate; B: Used from Vogler C, Rosenberg HS, Williams JC, et al. Electron microscopy in the diagnosis of lysosomal storage diseases. Am J Med Genet Suppl 1987;3:243-255, with permission.) |
mental retardation, seizures, recurrent infections, coarse facies, dysostosis multiplex and growth retardation, angiokeratoma corporis diffusum, and visceromegaly (73,77). The clinical course is variable. Most patients reach the second decade of life. The CNS, conjunctiva, muscle, skin and sweat gland epithelium, and peripheral blood lymphocytes all show granular lysosomal storage by EM (Figure 5-7). Diagnosis is based on demonstrating deficient alpha-l-fucosidase in leukocytes and fibroblasts. Some clinically normal individuals have low alpha-l-fucosidase levels in plasma.
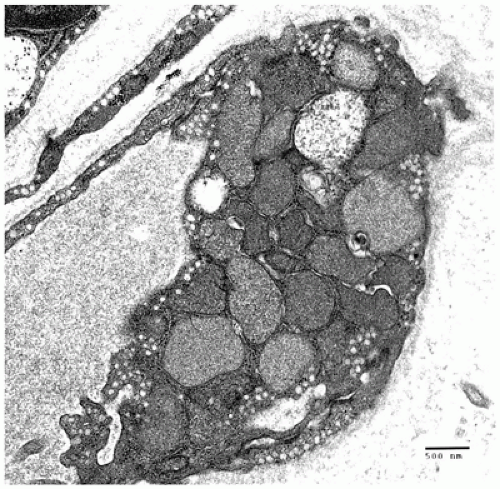 FIGURE 5-7 • Fucosidosis. Granular storage material distends the cytoplasm of this endomysial endothelial cell from a muscle biopsy of a 2-year-old girl with fucosidosis (Uranyl acetate, lead citrate). |
heparan, chondroitin, and keratan sulfate, with resultant storage of undegraded GAG in lysosomes in a variety of cell types (12). The clinical presentation is variable but may include facial dysmorphism (Figure 5-9A,B), bone/joint dysplasia (Figure 5-9C), hepatosplenomegaly, neurologic impairment, developmental regression, and short life span to mild clinical phenotype with normal life expectancy (Table 5-6) (87,88). All MPS are autosomal recessive except X-linked Hunter syndrome. Hurler (type I) and Hunter (type II) syndromes are the most common types. Scheie and Hurler-Scheie syndromes are subtypes of MPS I with a milder disease. Some infants, particularly with MPS VII, present with hydrops (Table 5-2).
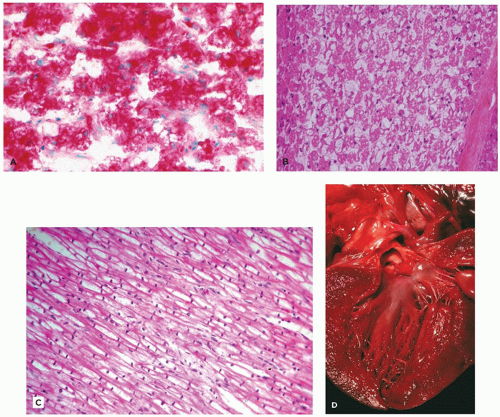 FIGURE 5-8 • Pompe disease (type II glycogen storage disease). A: Glycogen storage in Pompe disease is lysosomal (in contrast to glycogen storage in the other types of glycogen storage disease). The distended lysosomes also contain abundant acid phosphatase activity, which can be demonstrated histochemically, here in skeletal muscle by acid phosphatase staining (acid phosphatase stain). B: Vacuolar myopathy, though not specific for Pompe disease, is nonetheless characteristic and often striking in this LSD. The pale vacuoles in skeletal muscle fibers (representing glycogen storage) seen with H&E stain can be highlighted by PAS stain (not shown) (H&E). C: Histologically, cardiac myocytes are enlarged due to sarcoplasmic expansion by pale, often vacuolar material (H&E). D: Cardiac myocyte enlargement can lead to a hypertrophic gross appearance of myocardium, as seen in this image of the left ventricle from an infant who died with Pompe disease. |
nerve or cord compression can be manifestations of soft tissue involvement (Figure 5-9C, G, and H) (Table 5-6) (89). Vessel walls and heart valves are often affected with storage with resultant sclerosis (Figure 5-9E,F), and endocardial fibroelastosis may occur. Neurons store both GAG and gangliosides leading to formation of membranous cytoplasmic bodies, zebra bodies, and fibrillogranular storage material (Figure 5-9I). Neuronal loss and gliosis are seen in some patients, and meningeal storage may contribute to hydrocephalus.
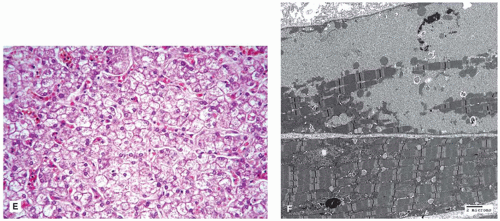 FIGURE 5-8 • (Continued) E: The histologic appearance of hepatocytes in Pompe disease is usually less striking than that of skeletal muscle. Hepatocytes are slightly enlarged with somewhat rarefied, vacuolar cytoplasm. Note the absence of glycogenated nuclei, which are typically not seen in the liver in Pompe disease but are observed in several other types of glycogen storage disease (H&E). F: Even though the primary defect in Pompe disease is lysosomal (in contrast to other glycogen storage diseases), glycogen accumulation is seen by ultrastructural analysis of skeletal muscle in the cytoplasm as well as within membrane-bound structures representing lysosomes. |
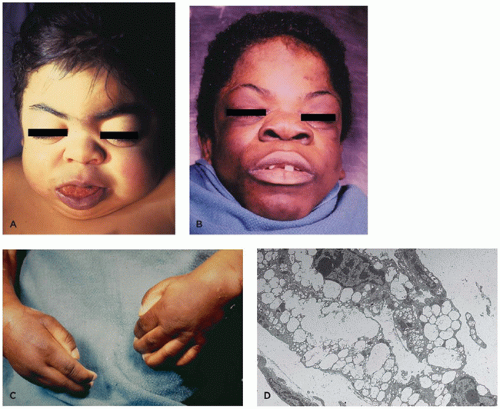 FIGURE 5-9 • Mucopolysaccharidosis. A, B: MPS patients have a characteristic facial appearance with coarse facial appearance, thick doughy skin, coarse hair, flattened midface, wide nasal bridge, and macroglossia, here seen in two children who died with MPS. C: The hands in MPS patients have joint stiffness and are held in a flexed position, a function of periarticular altered connective tissue and altered bone formation. D: In mucopolysaccharidosis, stored GAGs (previously called mucopolysaccharides) have the ultrastructural appearance of fine fibrillogranular material and clear membrane-bound vacuoles. Distinguishing different types of mucopolysaccharidosis based on ultrastructural morphologic characteristics is not possible. |
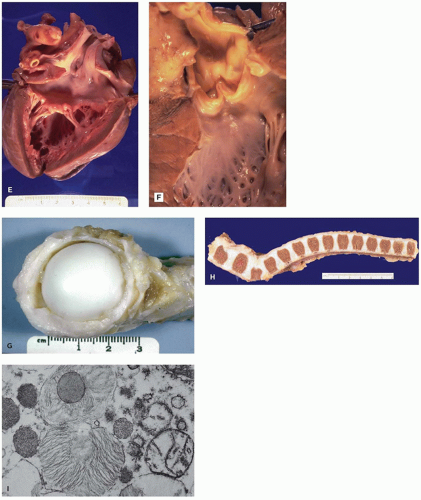 FIGURE 5-9 • (Continued) E, F: The heart in patients with MPS typically has thickened sclerotic valves, due to GAG storage in heart valve stromal cells and altered extracellular connective tissue in the valve. Endocardial thickening is also frequent. G, H: The femoral head of a patient from MPS shows articular synechiae and thick, poorly pliable periarticular connective tissue. These joint changes cause marked joint stiffness and make normal movement impossible. The vertebral column from an MPS patient shows characteristic anterior inferior breaking of the lower thoracic and upper lumbar areas caused by hypoplasia of the anterior superior aspect. This change results in the dorsal kyphosis or gibbus deformation often seen in MPS patients, and it is part of the widespread dysostosis multiplex. I: Though storage material in neurons can resemble that seen in other organs, it can also take the form of “zebra bodies,” as shown in this case of Hurler syndrome (D, I: Uranyl acetate, lead citrate). |
lysosomal storage. LM of thick sections of tissue prepared for EM is useful for identifying the multiple clear cytoplasmic vacuoles indicative of lysosomal storage. Enzyme assay of serum, leukocytes, or fibroblast culture provides definitive diagnosis, and carrier testing using DNA analysis is practical. Enzyme replacement therapy is now available for some forms (87,88).
TABLE 5-6 MUCOPOLYSACCHARIDOSES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Cholesterol and triglycerides in these cells can also be highlighted histochemically with the Schultz modification of the Lieberman-Burchard reaction. EM shows lipid droplets and membrane-bound angular cholesterol clefts in hepatocytes, Kupffer cells, fibroblasts, and macrophages (Figure 5-10D). The mucosa of the small intestine, particularly duodenum and ileum, is velvety yellow due to lamina propria storage. Adrenal glands are large, hard, and bright yellow, with dystrophic calcification and necrosis of the inner fasciculata and residual fetal cortex. Oligodendroglia, some neurons of the CNS, and Schwann cells of the peripheral nervous system contain lipid. Placental syncytiotrophoblasts may be affected. Demonstration of acid lipase deficiency in tissue, cultivated fibroblasts, or leukocytes confirms the diagnosis.
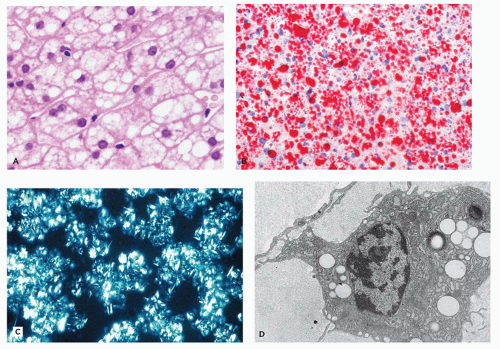 FIGURE 5-10 • Cholesterol ester storage disease. A: In cholesterol ester storage disease, there is widespread vacuolization of hepatocytes (H&E). B: Widespread cytoplasmic lipid can be demonstrated in frozen section analysis of liver tissue in cholesterol storage disease, here stained with oil red O. C: Hepatocellular cholesterol ester crystals are birefringent in frozen sections when viewed with polarized light. D: This conjunctival macrophage does not show needle-shaped clefts but does show many sharply demarcated electron-lucent vacuoles, some of which have peripheral osmiophilia, characteristic of lipid following fixation. (D: Uranyl acetate, lead citrate, Used from Vogler C, Rosenberg HS, Williams JC, et al. Electron microscopy in the diagnosis of lysosomal storage diseases. Am J Med Genet Suppl 1987;3:243-255, with permission.) |
synthesized lysosomal hydrolases for their appropriate targeting toward lysosomes by adding mannose-6-phosphate as a signal in the Golgi network (93). Phosphotransferase deficiency results in abnormal lysosomal enzyme transport. As a result, newly synthesized enzymes are secreted out of the cell instead of being transferred to lysosomes. This results in elevated plasma levels of lysosomal enzymes. The GNPTAB gene affected in ML II encodes the alpha- and beta-subunits of the hexameric enzyme complex, and the GNPTG gene affected in ML III encodes the gamma subunit.
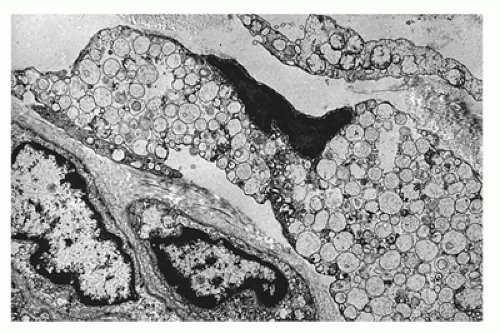 FIGURE 5-11 • I-cell disease. In I-cell disease, fibroblast cytoplasm is expanded by numerous membrane-bound vacuoles containing electron lucent to fibrillogranular material (Uranyl acetate, lead citrate). |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


