The Pancreas
Mariko Suchi, M.D., Ph.D.
ORGANOGENESIS AND EXOCRINE HISTOGENESIS
During week 4 of gestation, the ventral foregut gives rise to two pancreatic buds at the junction of the hepatic duct. The dorsal primordium develops in the mesentery. The ventral pancreatic bud rotates with the gut and fuses with the larger dorsal anlage and with the duodenum, and the fused primordia take up their normal position against the posterior abdominal wall in the concavity of the duodenum. The ventral bud gives rise to the uncinate process and the inferior portion of the head of the pancreas, and the dorsal primordium gives rise to the body, tail, and superior portion of the head (Figure 16-1). The duct of the dorsal pancreas opens more proximally into the duodenum and the ventral duct more distally, together with the common bile duct. Fusion of the primordia during the middle of week 6 of gestation leads to fusion of the duct systems. The duct of the ventral pancreas becomes the main pancreatic duct of Wirsung. The duct of the dorsal pancreas usually remains patent as the minor duct of Santorini.
During week 7 of gestation, simple, undifferentiated epithelial tubules grow into a loose mesenchyme. The epithelium rapidly forms a ramifying duct system, from which buds of cuboidal cells form the first recognizable acinar units by week
10; endocrine elements are also present by this time. The pancreas, from week 10 to term, continues to ramify and gives rise to exocrine and endocrine elements. Ductal elements, especially the centroacinar cells, can be hard to identify definitively from acinar and endocrine cells at this stage, but they express keratins 7 and 19 in addition to the keratins 8 and 18, which are found on acinar and endocrine cells after week 16 (1).
Complex yet organized gene regulatory networks governing pancreas development are reviewed by Arda et al. (2).
Two of the genes are noteworthy as they interact, induce, and are regulated by multiple other gene products and appear to be of vital importance to early pancreatic embryogenesis. They are PTF1A (pancreas transcription factor 1 α) and PDX1 (pancreas and duodenal homeobox 1). Very little is known about the embryologic control of pancreatic duct formations in humans.
Pancreatic exocrine development, like pulmonary alveolar maturation, is largely a postnatal event. The appearance of the pancreas at birth is one of underdeveloped acinar elements in a mesenchymal stroma (Figure 16-2). Acinar tissue increases rapidly after birth. Imrie et al. (3) showed that the ratio of acinar to connective tissue volume increases in a linear manner from 0.5 at week 32 to 2.0 at week 52 after conception. In an assessment of the morphologic maturation of an infant pancreas, the length of postnatal survival is more important than is the gestational age.
CONGENITAL ANOMALIES AND MALFORMATIONS
Agenesis and Hypoplasia
Agenesis of the pancreas refers to a complete absence of the gland, rather than a lack of the endocrine or exocrine portions alone. Recessive mutations in a distal enhancer of one of the organ development regulatory genes mentioned above, PTF1A, are the most common cause of isolated pancreatic agenesis. Rare cases of pancreatic agenesis result from PDX1 mutations. Congenitally absent pancreas may be associated with diaphragmatic hernia. Some infants have also lacked a gallbladder or intrahepatic bile ducts (4).
Functional pancreatic agenesis refers to organ failure without anatomic evidence of gland absence (5). A functionally hypoplastic pancreas may be familial and can also lead to exocrine and endocrine insufficiency. Severe pancreatic hypoplasia has been described in the Wolcott-Rallison syndrome (6).
Partial agenesis has been ascribed to failure of development of the dorsal or ventral primordium and may be familial
(7). More commonly, the pancreas is misshapen and described as short, stubby, or globular. These variants are not associated with hypofunction. Short pancreas may be an isolated finding or may be seen with complex congenital heart disease or as part of a wider malformation complex that includes congenital heart disease, multiple spleens, and intestinal malrotation. It has also been observed in complete trisomy 9 (8) and complete trisomy 22 syndromes.
Figure 16-3 illustrates a short pancreas in association with syndromic extrahepatic biliary atresia.
Figure 16-3 illustrates a short pancreas in association with syndromic extrahepatic biliary atresia.
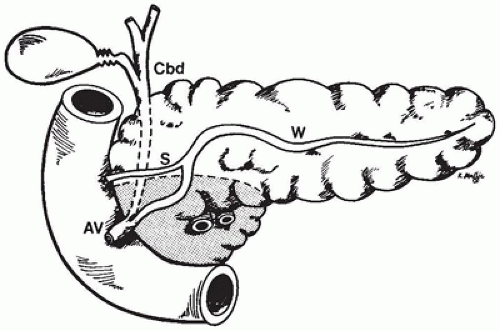 FIGURE 16-1 • Pancreas, ducts, and derivation. The pancreatic duct of Wirsung (W) drains most of the pancreas, joining the common bile duct (Cbd) proximal to the ampulla of Vater (AV). The accessory duct of Santorini (S) may drain part of the gland through a separate opening. A portion of the head and the uncinate lobe (stippled area) are derived from the ventral bud of the pancreas. |
Pancreatic Enlargement
Infants with Beckwith-Wiedemann syndrome have an enlarged pancreas. Marked fatty replacement of the exocrine portion with preservation of the islets is found in the lipomatous pseudohypertrophy of the Shwachman-Diamond syndrome.
Immune and nonimmune hydrops fetalis can lead to pancreatic enlargement through extramedullary hematopoiesis, and infiltration with leukemia can cause massive pancreatic enlargement (Figure 16-4). In congenital syphilis, pancreatomegaly is a consequence of extensive interstitial fibrosis and inflammation (9) (Figure 16-4).
Abnormalities of Position
The pancreas and the duodenum are retroperitoneal and separated from the posterior abdominal wall by an avascular plane. Abnormalities of fixation or position are often associated with left-sided diaphragmatic hernias. Partial situs inversus with normal cardiac situs but inversion of the abdominal viscera, including the pancreas, has been seen with annular pancreas (10).
Annular Pancreas
A ring of pancreatic tissue can encircle the second portion of the duodenum completely or partially (Figure 16-5).
Johnston described two forms, extramural and intramural
(11). In the extramural form, a flattened band of normal pancreatic tissue can be separated from the duodenum. A duct originating anteriorly runs around the duodenum to join the main pancreatic duct. In the intramural form, ectopic pancreatic tissue is located within the duodenal wall, and small ducts drain directly into the duodenum.
Children and neonates with annular pancreas usually present with duodenal obstruction. Duodenoduodenostomy is the appropriate treatment, with good postsurgical outcome
(12). As compared to the adult patients with annular pancreas, children with this anomaly frequently have associated chromosomal anomalies and major congenital malformations
(13). The most frequent chromosomal anomaly is trisomy 21. Congenital malformations associated with annular pancreas include tracheoesophageal fistula, Meckel diverticulum, absence of the gallbladder, and imperforate anus. Annular pancreas in a mother and three of her four children and documentation of other familial instances suggest an autosomal dominant inheritance or involvement of an autosomal recessive sex-influenced gene (14).
Annular pancreas may present in the fetus with polyhydramnios or in the neonate with bile-stained vomiting if the constriction is below the ampulla. In older children and adults, it may become symptomatic if duodenal ulceration or pancreatitis develops.
Ectopic Pancreas
Ectopic (heterotopic) pancreas is widely distributed, largely within the gastrointestinal tract. This condition has been discovered in 2% to 15% of all autopsies. Pancreatic tissue is most frequently found in the wall of the stomach, duodenum, or jejunum (15). It is not unusual to find a nodule in the stomach consisting of a centrally ulcerated pit with localized thickening of the gastric wall (Figure 16-6). Ectopic mediastinal pancreas is rare. Pancreatic tissue can be identified within bronchopulmonary foregut malformation (16).
Three types of ectopic pancreatic tissue have been described. The first is similar to normal pancreas showing a full complement of acinar, ductal, and islet constituents with a normal distribution of all cell types as demonstrated by immunohisto chemistry.
The second is characterized by incomplete lobular arrangement, few acini, many ducts, and an absence of endocrine elements. In the third type of ectopic pancreas, only proliferating ducts are present, without acinar or endocrine elements. This form is usually interpreted as an “adenomyoma” or “myoepithelial hamartoma” of the bowel wall (17).
Pancreatic tissue can be seen in the hilum of the liver or within the liver substance. Although on occasion a metaplastic process has been suggested for microscopic focus of exocrine pancreatic tissue in a posthepatitic cirrhotic liver, it is unlikely to be metaplastic when endocrine cells are seen. The pancreas has also been described in the omentum, mesentery,
Meckel diverticulum, vitelline duct, and umbilicus. Ectopic pancreas has been associated with duplication cysts of the gut and has been reported in fallopian tubes, abdominal lymph nodes, and adjacent to the thyroid (18). Intrasplenic islands of pancreas are found in trisomy 13 to 15 syndrome (19).
Although heterotopic pancreas is often an incidental finding, it may present clinically as peptic ulceration, massive hemorrhage, biliary obstruction, cholecystitis, pyloric obstruction, intestinal obstruction, intussusception, or cystic degeneration (15). When complicated by pancreatitis, it can present as a
mesenteric mass (20). Hyperinsulinism associated with islet cell adenomatous hyperplasia (focal hyperinsulinism) in the ectopic pancreas has been described (21). Solid pseudopapillary neoplasms have been identified in ectopic pancreas (22).
mesenteric mass (20). Hyperinsulinism associated with islet cell adenomatous hyperplasia (focal hyperinsulinism) in the ectopic pancreas has been described (21). Solid pseudopapillary neoplasms have been identified in ectopic pancreas (22).
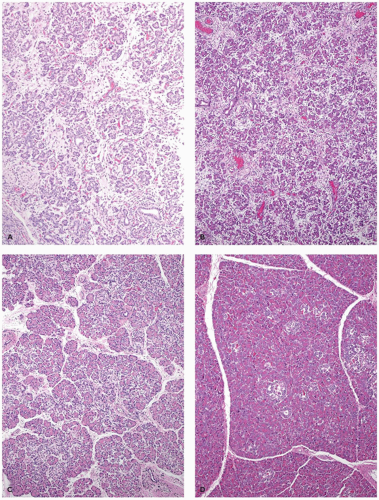 FIGURE 16-2 • Pancreatic development during gestation. A: At weeks 12 to 13 of gestation, the pancreas shows ducts and small lobules of acini in a loose mesenchymal stroma. Endocrine cells are demonstrable. (Hematoxylin and eosin stain, original magnification ×155.) B: At week 17 of gestation, lobules of acini and islets are more clearly visible. C: By week 40 of gestation (in a child who lived 1 day after birth), the endocrine elements lie centrally within the stalk of the lobule, and undeveloped acinar tissue is located peripherally. D: At the age of 22 months, the remarkable development of acinar tissue is obvious. (B-D: Hematoxylin and eosin stain, original magnification ×100.) |
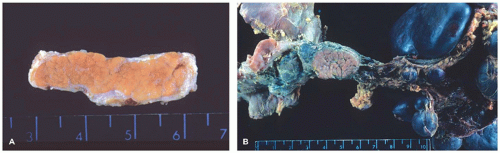 FIGURE 16-3 • Small pancreas. A: Hypoplastic pancreas (2.7 g, 3 cm long) from a 5-monthold child with probable tyrosinemia. B: Short pancreas (3 cm long) from a 2.5-year-old child with multiple spleens, intestinal malrotation, left bronchopulmonary isomerism, and extrahepatic biliary atresia (heterotaxy). |
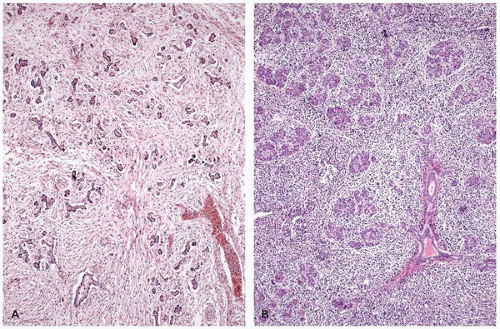 FIGURE 16-4 • Large pancreas. A: Congenital syphilis. An extensive, fine fibrosis distorts the pancreas. B: Congenital leukemia. Acinar elements are widely separated by the leukemic infiltrate, which is granulocytic in this instance. (A, B: Hematoxylin and eosin stain, original magnification ×100.) |
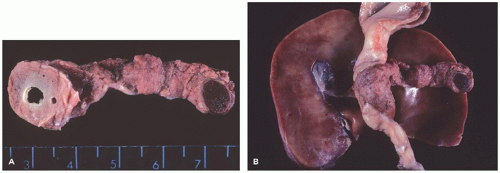 FIGURE 16-5 • Annular pancreas. A, B: An annular pancreas completely surrounds the second portion of the duodenum. An accessory spleen is present within the tail of the pancreas (trisomy 6). |
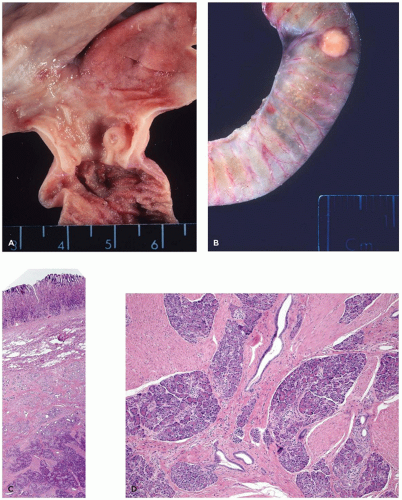 FIGURE 16-6 • Ectopic pancreas. A: Umbilicated mucosal nodule of the pyloric area. B: A jejunal subserosal nodule. C: An ectopic intramural (gastric) pancreas has ducts, exocrine acini, and endocrine component. D: Higher magnification of C. Islands of pancreatic tissue are separated by smooth muscle bundles. (C, D: Hematoxylin and eosin stain, original magnification, ×25 and ×100, respectively.) |
Pancreatic Cysts
Due to increased availability of advanced cross-sectional imaging techniques, congenital and acquired pancreatic cysts are more frequently encountered. They are more likely asymptomatic. True cysts have epithelial lining such as cuboidal epithelium, metaplastic stratified squamous epithelium, and ciliated columnar epithelium (23). “Dermoid cysts” have been also described in children, among which some are mature cystic teratoma upon reviewing the literature. Simple epithelium-lined cysts may be solitary and unassociated with cystic diseases of other organs. On the other hand, the cysts may be associated with a trisomy (19) (Figure 16-7), tuberous sclerosis, von Hippel-Lindau disease,
and Beckwith-Wiedemann syndrome (24) (Figure 16-8). The pancreatic lesions in Beckwith-Wiedemann syndrome and von Hippel-Lindau disease are described separately later in the chapter.
and Beckwith-Wiedemann syndrome (24) (Figure 16-8). The pancreatic lesions in Beckwith-Wiedemann syndrome and von Hippel-Lindau disease are described separately later in the chapter.
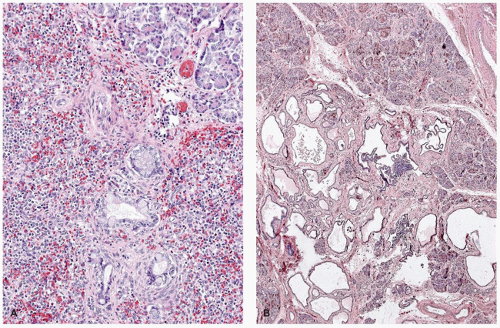 FIGURE 16-7 • Trisomies. A: Trisomy 13. Poorly circumscribed areas of splenic stroma extend into the pancreas, entrapping pancreatic ducts with mucoid lining cells. (Hematoxylin and eosin stain, original magnification ×200.) B: Trisomy 18. Numerous pancreatic cysts lined by duct-type epithelium are scattered. (Hematoxylin and eosin stain with immunohistochemistry for neuron-specific enolase, original magnification ×50.) |
In contrast to true cysts, a pancreatic pseudocyst is a collection of pancreatic secretions enclosed in fibrous tissue layer without an epithelial lining. Pseudocysts can arise as a complication of either chronic or acute pancreatitis due to any cause including infections, drugs, genetic predisposition, abdominal trauma, previous surgery, and structural pancreatic duct anomaly. Management options include observation and percutaneous or internal drainage (25). Lymphatic malformation (26) (Figure 16-9) may present as “pancreatic cysts.” Pancreatic cysts in cystic fibrosis are pseudocysts and will be discussed separately.
Pancreatic Anomalies in Nonmotile Ciliopathies
Cilia are hairlike structures or organelles that extend from the surface of nearly all mammalian cells. There are two types of cilia, motile and nonmotile. Nonmotile cilia are also known as primary cilia, which play a crucial role as sensory cellular antennae during embryogenesis, guiding developing cells to their ultimate fate. A number of heterogeneous phenotypes and syndromes that manifest with a variety of abnormalities in multiple organ systems including the pancreas have been historically described. The overlapping natures of the syndromes resulted in confusion in diagnostic criteria and classification. Some of these syndromes have been classified as ciliopathies (27) including, but not limited to, autosomal recessive polycystic kidney disease, autosomal dominant polycystic kidney disease, renal-hepatic-pancreatic dysplasias, Meckel-Gruber syndrome, Joubert syndrome-related disorder, and Jeune syndrome. As cilia are present in nearly every cell in the body and mutations of genes encoding ciliary proteins affect multiple organs, including the kidneys, liver, pancreas, retina, central nervous system, and skeletal system, genetic defects will manifest with multiorgan involvement.
The prototype is autosomal dominant kidney disease. This disorder has cysts in the kidney and liver. But cases in this category have been reported to have pancreatic cystic lesions (eFigure 16-1). By ultrasonography, pancreatic cysts are detected in 9% of autosomal dominant polycystic kidney disease patients over 30 years of age.
Renal-hepatic-pancreatic dysplasia 1 (also known as Ivemark II syndrome) is caused by homozygous or compound heterozygous mutation in nephrocystin 3 gene, NPHP3, on chromosome 3q22. Pancreatic findings include fibrosis, epithelium-lined cysts, dilated pancreatic ducts, and decrease in parenchymal tissue (28). Endocrine function varies from normal to diabetes mellitus. Another gene, never in mitosis gene A-related kinase 8 gene (NEK8), whose homozygous mutation causes another renal-hepatic-pancreatic dysplasia has been discovered recently (renal-hepatic-pancreatic dysplasia 2).
Meckel syndrome, also known as Meckel-Gruber syndrome, shows extensive clinical variability and may include
pancreatic cystic lesions (eFigure 16-1). This variability is understandable because there are currently 11 types based on the causative gene mutations.
pancreatic cystic lesions (eFigure 16-1). This variability is understandable because there are currently 11 types based on the causative gene mutations.
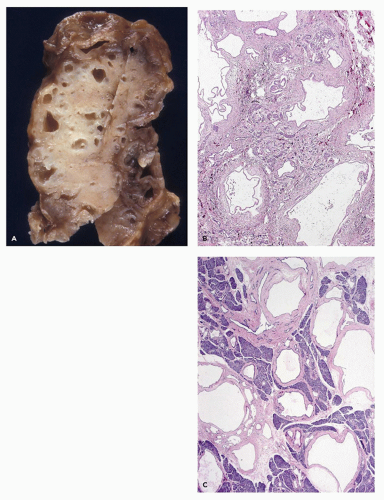 FIGURE 16-8 • Pancreatic cysts. A: A large pancreas (120 g) presented as an abdominal mass in a newborn baby with Beckwith-Wiedemann syndrome. Multiple cystic spaces are seen on cut surfaces. (Color version of Copyright 1990 from Beckwith-Wiedemann syndrome with unusual hepatic and pancreatic features: A case expanding the phenotype by Steigman CK, Uri AK, Chatten J, et al. Pediatr Pathol 1990;10:593. Reproduced with permission of Taylor & Francis Group, LLC., http://www.taylorandfrancis.com.) B: Microphotograph of (A). No normal pancreatic tissue is identified. Numerous ectatic ducts, clusters of endocrine cells, and few acini are in loose fibrous connective tissue. (Hematoxylin and eosin stain, original magnification ×50.) C: 15-year-old child with oral-facial-digital syndrome type I. Cystically dilated pancreatic ducts with periductal fibrosis. (Hematoxylin and eosin, original magnification ×25.) |
Variations in the Pancreatic Ducts
After the dorsal and ventral pancreatic anlage fuse, the dorsal duct becomes the accessory duct of Santorini, and the ventral duct becomes the main draining duct of the pancreas, the duct of Wirsung. Fusion can occur at a single point proximally or at two points, both proximally and more distally (29). The normal architecture varies widely. The dorsal duct may regress completely, or the dorsal and ventral ducts may not communicate at all (pancreas divisum).
In pancreas divisum, the pancreas is separated into two portions. The pancreatic ducts fail to fuse, and the dorsal duct of Santorini drains most of the gland through the minor
papilla. The ventral duct of Wirsung drains only the smaller pancreatic remnant. Because of the recent advances in endoscopic retrograde cholangiopancreatography and magnetic resonance cholangiopancreatography, pancreas divisum is diagnosed with greater frequency. Some investigators believe that pancreas divisum is clinically significant, whereas others are more skeptical.
papilla. The ventral duct of Wirsung drains only the smaller pancreatic remnant. Because of the recent advances in endoscopic retrograde cholangiopancreatography and magnetic resonance cholangiopancreatography, pancreas divisum is diagnosed with greater frequency. Some investigators believe that pancreas divisum is clinically significant, whereas others are more skeptical.
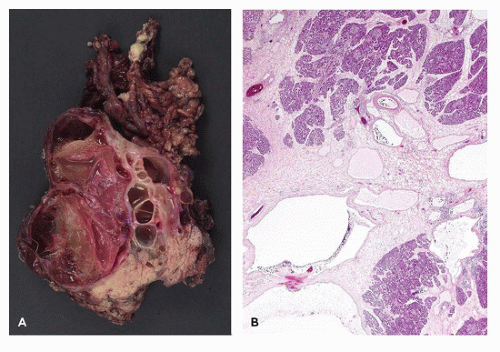 FIGURE 16-9 • Lymphatic malformation. A: Lymphatic malformation presented as a cystic pancreatic mass in a 12-yearold child. (Courtesy of Pierre Russo, M.D., Philadelphia, Pennsylvania.) B: Lymphatic channels of variable sizes are embedded within the pancreatic lobular septa and parenchyma. A small number of mononuclear cell infiltrates are present in the walls. (Hematoxylin and eosin stain, original magnification ×25.) |
The anatomy of the pancreaticobiliary junction varies noticeably. The pancreatic duct can join the common bile duct within the duodenal wall, or it can enter the duodenum separately. The common channel can be short or long. If it is longer than 2 cm, the ducts join outside the duodenal wall.
Stenosis of the ampulla may present with pancreatitis, but in the newborn, it more commonly manifests as bile duct perforation.
Familial cases of pancreaticobiliary anomaly have been reported (30).
Pancreatic Pathology in Trisomies and Triploidy
Lesions of the pancreas characteristic of trisomy 13 were illustrated long before the chromosomal defect was identified. Hashida et al. (19) demonstrated the multiple, poorly demarcated aggregates of splenic tissue in the tail and the body of the pancreas (Figure 16-7). These splenic islands contain pancreatic acini, islets, and knots of ducts lined by tall columnar epithelium rich in goblet cells. Microcystic changes may be focal or quite widespread with inspissations, features that suggest a duct obstruction as the pathogenesis. Ectopic splenic tissue in the gastric fundus and upper pole of the kidney has contained pancreatic elements.
The pancreas in trisomy 18 often exhibits lobular fibrosis (19) and focal fibrotic nodules in which clusters of ducts and atrophic acini are found. An area of serous cystadenomatous changes with back-to-back cysts is seen less frequently (Figure 16-7). These cysts along with inflammatory aggregates suggest an obstructive etiology. Ectopic pancreas in the ileum was reported in a case of trisomy 18 (31).
Annular pancreas occurs in as many as 8% of infants with trisomy 21. Pancreaticobiliary duct anomalies may accompany annular pancreas (32). A short pancreas has been described in trisomy 22. A report of three cases of complete trisomy 9 that survived beyond the first trimester (a rare occurrence) described agenesis or underdevelopment of the body and tail of the pancreas in each of them (8).
Pancreatic anomalies, including agenesis, are described in infants with triploidy. Marked enlargement of the somatostatin-producing D cells has been reported in triploid fetuses (33).
EXOCRINE PANCREAS
Functional Development
Exocrine function of developing pancreas varies significantly among specific enzymes. Immunohistochemical analysis of the fetal pancreas detects pancreatic secretory trypsin inhibitor in developing buds of the pancreas during the 8th gestational week, and proteinases such as trypsin, chymotrypsin, and elastase I are observed in acinar cells during the 14th week of gestation. Morphologically, small zymogen-like granules are present in a few young acinar cells at 12 weeks. During the 12 to 19 fetal week period, there is a gradual increase in the number of mature zymogen granules (34). By week 20 of gestation, basal round granules predominate, and the complex basolateral cell interdigitations are established (eFigure 16-2). These timings coincide with the levels of proteinase activity and immunohistochemical reaction patterns. After 32 weeks of gestation, both preterm and term infants have substantial proteolytic enzyme levels. Chymotrypsin activity is about 50 % to 60% of the level found in older children. Trypsin activity, the highest of all enzymes, reaches
90% to 100% of children older than 2 years of age (35). In the adult, pancreozymin is responsible for pancreatic enzyme release, and secretin promotes fluid and electrolyte release. The pancreas of a neonate is unresponsive to exogenous pancreatozymin or secretin before 1 month of age and not fully responsive before 2 years of age.
90% to 100% of children older than 2 years of age (35). In the adult, pancreozymin is responsible for pancreatic enzyme release, and secretin promotes fluid and electrolyte release. The pancreas of a neonate is unresponsive to exogenous pancreatozymin or secretin before 1 month of age and not fully responsive before 2 years of age.
For lipase and amylase secretion, the pancreas appears to mature through a different time course. Throughout the fetal period studied, mRNA of lipase is shown to be consistently and significantly lower than trypsinogen mRNA level. By immunohistochemistry, while trypsinogen and chymotrypsinogen are observed uniformly in all acinar cells as soon as they appear at 16 weeks of gestation, lipase is detected at 21 weeks in a few acini dispersed in the pancreas and then spreads out progressively to be present in all the acini (36). The duodenal fluid of newborns and infants, nevertheless, contains low levels (5% to 10% of adult values) of lipase, and infants are said to have physiologic steatorrhea (37).
Earlier studies claim that amylase could be detected in the fetal pancreas beginning in the 22nd week of gestation. However, a later study by Fukuyama et al. on fetal pancreas using immunohistochemistry and studies involving postnatal enzyme activity measurements of the duodenal fluid show lack of amylase in the fetal pancreas and early postnatal pancreas of up to 1 month of age (35). The amylase level in the duodenum increases slowly during the first 1 to 2 years of life.
Abnormalities of the Exocrine Pancreas
Isolated Enzyme Deficiencies
Rare pediatric cases of isolated pancreatic exocrine enzyme deficiencies were reported in the 1960s and in the years up to the early 1990s. These patients were without known causes of pancreatic insufficiency such as pancreatitis, tumor, and congenital syndromes. The deficiency is determined by enzymatic activity assays in pancreatic juices. Deficient enzymes include lipase, colipase, and trypsinogen. The pancreatic morphology is not described in these reports except for one case of trypsinogen deficiency whose pancreas showed “immature acini” and lack of zymogen granules. Mutational analysis of the gene encoding pancreatic lipase performed on six members of a consanguineous family identified a missense mutation that shows segregation between the genotype state and the serum pancreatic lipase activity.
Enteropeptidase is a serine protease that activates the pancreatic proenzyme trypsinogen, which, in turn, releases active digestive enzymes from their inactive pancreatic precursors. Therefore, congenital enteropeptidase deficiency may mimic cystic fibrosis manifesting with failure to thrive and diarrhea. But this enzyme is produced from the intestinal brush border in the proximal small intestine, not from the pancreas. Nonsense mutations and a frame shift mutation have been identified in the proenteropeptidase gene.
The maturation of amylase production is normally delayed, and the adult level of amylase activity is reached at the age of 18 months to 2 years. Therefore, alleged cases of isolated amylase deficiency in young children are controversial. Rare adult cases of selective deficiency of pancreatic amylase have been described.
Diseases that Result in Replacement of Exocrine Acini by Adipose Tissue
When the shape of the pancreas is grossly preserved but the exocrine acini are largely replaced by adipose tissue, the condition is termed lipomatous atrophy, lipomatosis, or, when extensive, lipomatous pseudohypertrophy. The pancreas shows relatively preserved endocrine components including islets of Langerhans and scattered small but well-preserved exocrine acinar elements. These acini do not show signs of injury. No fibrosis or fat necrosis is seen. Lipomatosis has been described in a number of pathologic processes (38) including Bannayan syndrome, Shwachman-Diamond syndrome, Johanson-Blizzard syndrome, and rare proteolytic and lipolytic enzyme deficiencies.
Of note, focal replacement of the exocrine pancreas with mature fat is a relatively common histologic change. It usually correlates directly with BMI and presence of diabetes mellitus.
Shwachman-Diamond Syndrome
With an incidence estimated at 1 in 200,000 births, the Shwachman-Diamond syndrome (39) is the most common cause of pancreatic insufficiency after cystic fibrosis. Pancreatic exocrine insufficiency is accompanied by growth restriction, short stature, bone marrow depression with neutropenia, and skeletal changes, predominantly metaphyseal dysostosis (40). Cases with anal atresia, Hirschsprung disease, and possibly asphyxiating thoracic dystrophy have been described (41). The marrow dysfunction, neutropenia in most instances, may be fixed or cyclical and is associated with the development of myelodysplasia and later leukemias, usually acute myelogenous leukemia. The pancreatic insufficiency may be profound and appears shortly after birth; less often, it is mild and improves and normalizes with age in about half of these patients. Replacement of the bulk of the pancreas by fatty tissue gives the appearance of a lipomatous pseudohypertrophy (42). Acinar tissue is absent, without scarring or fibrosis, and the pancreatic ducts and endocrine elements are preserved (Figure 16-10). The characteristic lipomatous pancreas can be demonstrated by magnetic resonance imaging.
Shwachman-Diamond syndrome is an autosomal recessive disorder. The gene involved (SBDS) resides at 7q11. Its 1.6-kb transcript encodes a predicted protein of 250 amino acids. A pseudogene copy (SBDSP) with 97% nucleotide sequence identity is in a locally duplicated genomic segment. Recurring mutations resulting from gene conversion (substitution of genetic material from another gene) were found in 89% of unrelated individuals of Shwachman-Diamond syndrome. The converted segments include pseudogene-like sequence changes that result in protein truncation. In a study of 23 unrelated patients, molecular genetic and hematologic evaluations demonstrated a poor genotype/phenotype correlation (43).
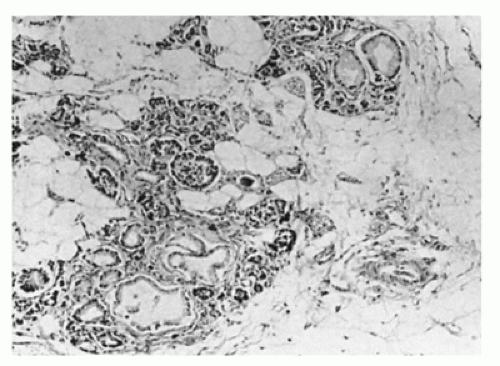 FIGURE 16-10 • Exocrine atrophy in Shwachman-Diamond syndrome. The acinar elements are almost completely replaced by fat (lipomatous pseudohypertrophy), with only small aggregates of endocrine tissue left around ducts. (Hematoxylin and eosin stain, original magnification ×120.) |
Johanson-Blizzard Syndrome
First reported in 1971 (44), the Johanson-Blizzard syndrome comprises congenital aplasia of the alae nasi, deafness, hypothyroidism, dwarfism, absence of permanent teeth, and malabsorption resulting from pancreatic insufficiency. Subsequent reports described urogenital abnormalities and imperforate anus associated with this syndrome. Postmortem examination of the pancreas reveals a total absence of acini with complete replacement of the pancreas by adipose tissue and a few remaining islets with connective tissue around the ducts (45). In contrast, an autopsy of a 4-year-old patient showed a pancreas consisting only of the head with immature-appearing acinar cells, centrally placed islets, periductal fibrosis, and ductular squamous metaplasia, but without significant fat replacement (46). A report regarding specimens from 2 fetuses (21 and 34 weeks of gestation) and a newborn baby revealed acinar tissue loss that increased with gestational age, accompanied by inflammatory infiltrates (47). There was no evidence of increased apoptosis in acinar cells by TUNEL assay, suggesting that the pancreatic defect may not be due to perturbed acinar development, but may rather be gradual destruction of previously formed acinar cells. Mutations in the gene UBR1 have been detected in affected individuals from 12 of 13 families (47). UBR1 encodes one of E3 ubiquitin ligases of the N-end rule pathway, a conserved proteolytic system. The UBR1 protein substrate, presumably impaired degradation of which causes Johanson-Blizzard syndrome, is not yet known. Using antibody to UBR1 and immunofluorescence microscopy, no UBR1 protein was observed in the pancreas from individuals with Johanson-Blizzard syndrome, while it is readily detectable in the cytosol of acinar cells in the control pancreas. Immunofluorescence pattern for trypsinogen indicated that there was no primary defect of zymogen synthesis. Hypoplasia of the exocrine pancreas in association with facial anomalies, micrognathia, posterior cleft palate, and dental hypoplasia has been referred to as the Donlan syndrome, but overlap with the Johanson-Blizzard syndrome seems likely (48).
Pearson Marrow-Pancreas Syndrome
Pearson et al. (49) described a syndrome characterized by pancreatic insufficiency, refractory sideroblastic anemia, and variable neutropenia in which the marrow is normocellular but the cells are vacuolated. A number of mitochondrial DNA deletions have been reported in affected individuals. The pancreatic pathologic features differ from those of the Shwachman-Diamond or Johanson-Blizzard syndrome in that acinar atrophy with fibrosis, not lipomatosis, is present (49,50).
A neonate with Pearson syndrome and diabetes mellitus (50) suggests a connection with Kearns-Sayre syndrome. The features of Kearns-Sayre syndrome, a mitochondrial myopathy, may develop later in life in patients who survive the early manifestations of Pearson syndrome. It has been postulated that a phenotypic shift from a predominantly hematopoietic disorder (Pearson syndrome) to a disease with muscle dysfunction (mitochondrial myopathy) and an eventual evolution to a full picture of Kearns-Sayre syndrome depends on the distribution of deleted mitochondrial DNA (51).
Neonatal Hemochromatosis
Neonatal hemochromatosis is unrelated to adult-type hereditary hemochromatosis. In neonatal hemochromatosis, hemosiderin deposition in the pancreatic acini is massive. The islets are generally spared, at least in the early stages (52) (Figure 16-11). Nevertheless, one case of neonatal diabetes due to neonatal hemochromatosis has been reported. Islets have been described to be increased in size in some cases, but the illustrations are less convincing. Acinar iron deposition in the absence of reticuloendothelial iron deposition is not pathognomonic of hemochromatosis; it was also seen in some control cases (53). Two siblings with a neonatal hemochromatosis phenotype that included pancreatic iron deposition also had hypertelorism and trichomalacia, a trichohepatoenteric syndrome (54).
Cystic Fibrosis
Fanconi et al. in 1936 and then Anderson in 1938, while investigating causes of malabsorption, discovered in some of their patients a condition they termed cystic fibrosis of the pancreas. Recognition of the pancreatic lesion preceded the clinical delineation of the disease. Farber (55) proposed mucous plugging of all secretory glands (mucoviscidosis) to be the pathogenetic key in 1944.
Cystic fibrosis is caused by defects in the cystic fibrosis conductance regulator gene (CFTR), localized to 7q31.2. Pancreatic insufficiency is not obligatory, and 15% of patients are clinically pancreas “sufficient.” The degree of pancreatic disease varies widely at any age, although the disease is progressive and leads to increasingly more severe changes with time. The final stage of cystic fibrosis is characterized by obstruction of the pancreatic ducts by viscous secretion leading to complete acinar atrophy accompanied by fibrosis and lipomatosis (56,57).
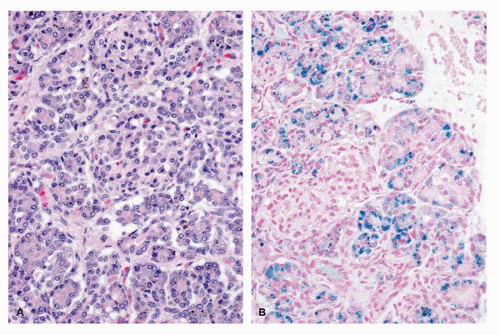 FIGURE 16-11 • Neonatal hemochromatosis. A: Exocrine acinar cells contain brown refractile pigment. (Hematoxylin and eosin stain, original magnification ×400.) B: The islets are devoid of iron, but acinar lobules have a marked intracytoplasmic iron deposition. (Prussian blue stain, original magnification ×400.) |
The tissue alteration can be recognized even before 40 weeks of gestation (3,57,58). The ratio of acinar to connective tissue volume is 0.5 at 32 weeks after conception, increasing to 2.0 at 52 weeks in normal controls (3). In the pancreas, the ratio of acinar to connective tissue, low to begin with, decreases from 0.5 at 35 weeks to 0.3 at 52 weeks after conception. Further degeneration of exocrine tissue supervenes postnatally.
The earliest visible lesions are eosinophilic concretions in acini and ductules, which may lead to acinar or ductular dilation and flattening of the lining epithelium (56). These concretions generally stain with periodic acid-Schiff (PAS) and contain calcium. The changes may be focal in preterm infants and in mildly affected cases. Postnatal acceleration of the pathogenetic train of inspissation, obstruction, dilation, epithelial damage, atrophy, cell destruction, and fibrosis with minimal inflammation leads to progressive acinar loss with replacement by fibrous tissue (Figure 16-12; eFigure 16-3) and adipose tissue.
Cystic fibrosis in the older child can be associated with large single or multilocular cysts (59). The pancreatic pseudocyst, caused by rupture of a duct into the lesser sac or abdominal cavity, consists of a fibroinflammatory wall around an autodigested cavity. In addition to cystic fibrosis, pseudocysts are caused by trauma, surgery, or inflammation (pancreatitis). The fibrous wall of the cyst has no epithelial lining.
Even though acinar tissue may disappear, islet tissue is preserved until very late. The endocrine changes in cystic fibrosis are considered later in this chapter.
Inspissation and Other Changes of Pancreatic Ducts
Baggenstoss (60) described the widespread inspissation of secretions in centroacinar cell-lined ductules in patients with uremia. The finding is not restricted to uremia but is also seen in children dying with acidosis, dehydration, cardiac failure, or sepsis. Inspissation may also be seen in the pancreas of children who have experienced prolonged hyperalimentation, but they usually have many of the other conditions listed.
Striking oncocytic changes of the ducts and centroacinar cells have been observed in an infant with mitochondrial myopathy, lactic acidosis, and “ragged red” muscle fibers (MELAR syndrome). Centroacinar hyperplasia of an impressive degree was observed in a young adult with HIV infection (eFigure 16-4).
Pancreatitis in Childhood
In contrast to pancreatitis in adulthood, which is frequently related to alcohol abuse, the common causes of pancreatitis in children are trauma, multisystemic diseases, medications, infections, and biliary tract diseases. What etiologies are included in the “multisystem” category vary among the studies (61,62). Pancreatitis can also be seen in children with branched-chain organic acidemias, methylmalonic acidemia, isovaleric acidemia, and maple syrup urine disease (63). When all causes are excluded and the etiology remains undetermined, the pancreatitis has been traditionally labeled idiopathic. This group accounts for 8% to 34% of cases and leads the “cause” of childhood pancreatitis in some series (61). Except for patients with cystic fibrosis, hereditary pancreatitis, and pancreatitis secondary to congenital structural, syndromic, or metabolic abnormalities, most children have a single, self-limited episode of pancreatitis, and few cases progress to chronicity (62). On the other hand, children with recurrent or so-called idiopathic chronic pancreatitis may possess mutations and sequence variations in modifier genes as discussed below.
Acute pancreatitis of any cause varies from interstitial edema to necrotizing hemorrhagic inflammation depending on the severity and time point of the process. Fat necrosis is the characteristic finding and results from sequential
reactions including release of fatty acids from triglyceride esters by lipase and combination of the fatty acids with calcium (saponification). The formed insoluble salts produce grossly visible chalky white to yellow areas. Microscopically, the outlines of degenerated fat cells with basophilic calcium deposits are seen along with acute inflammatory infiltrate (Figure 16-13A).
reactions including release of fatty acids from triglyceride esters by lipase and combination of the fatty acids with calcium (saponification). The formed insoluble salts produce grossly visible chalky white to yellow areas. Microscopically, the outlines of degenerated fat cells with basophilic calcium deposits are seen along with acute inflammatory infiltrate (Figure 16-13A).
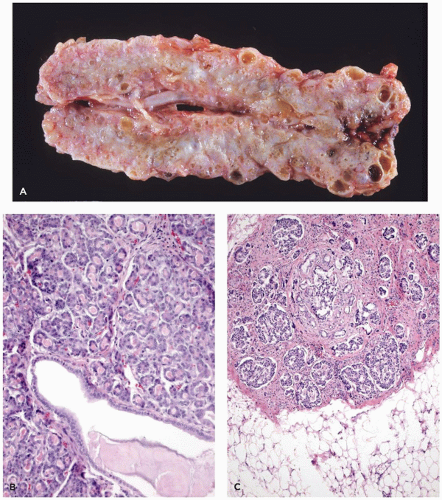 FIGURE 16-12 • Cystic fibrosis. A: The pancreas of this 12-year-old child is lipomatous with scattered cysts. B: An 8-year-old child with a strong family history of cystic fibrosis was asymptomatic and, at autopsy, had only cystic dilation with inspissation of ducts and acini with minimal fibrosis. (Hematoxylin and eosin stain, original magnification ×200.) C: The pancreas of a 10-year-old child shows advanced acinar atrophy, fatty replacement and periductal endocrine overgrowth. (Hematoxylin and eosin stain, original magnification ×100.) |
Recurrent pancreatitis and chronic pancreatitis produce fibroinflammatory changes of the parenchyma (64). The pancreas shows extensive fibrosis, acinar atrophy characterized by reduced number and size of acini, and variable dilatation of ducts with calcification (Figure 16-14). The endocrine islets are generally relatively spared and embedded in fibrotic tissue. They may appear fused and enlarged, but in end-stage disease, they eventually disappear.
Traumatic Pancreatitis
Blunt trauma is recognized as a cause of immediate or delayed pancreatitis in children (Figure 16-13A) and an important cause of pancreatic pseudocyst. A pancreatic pseudocyst may occur as the result of child abuse, but bicycle injuries are the most common cause in children.
Infectious Pancreatitis
The pancreas may be the seat of any disseminated infection, such as herpes simplex, cytomegalovirus infection (Figure 16-13B), or bacterial sepsis, but this may not result in clinically relevant pancreatitis (65,66). In some instances, the late consequences of a previous infectious pancreatitis are serious.
Rubella may cause an interstitial pancreatitis as part of the expanded congenital rubella syndrome, and pancreatic
insufficiency or even diabetes mellitus can ensue (67). Mumps is a classic cause of pancreatitis in late childhood. Diffuse necrosis and pseudocyst formation has been reported in a surgical specimen (66). Coxsackievirus may selectively involve the exocrine or endocrine components of the pancreas with parenchymal hemorrhage (65,66). Parainfluenza 3 pancreatitis, confirmed by immunohistochemistry, produced multinucleated giant cells in the pancreas of a child with severe combined immunodeficiency. Varicella-zoster infection (chicken pox) resulted in pancreatitis in individuals succumbed to the disease. The postmortem examination revealed focal pancreatic necrosis (66). Chronic coxsackievirus B interstitial pancreatitis in the absence of a meningoencephalomyocarditis in a child with α1-antitrypsin deficiency has been reported (68). Pancreatitis is a well-recognized consequence of bone marrow transplantation. Adenovirus infection may be the causative agent in some cases. In most cases of pancreatitis in children with acquired immunodeficiency syndrome (AIDS), the disease is mild and reflects systemic disease status. The incidence of opportunistic infections is low, although nonspecific inflammatory changes such as focal lymphoplasmacytic aggregates are common (69). Escherichia coli pancreatitis usually accompanies septicemia. Congenital syphilis causes a pancreatitis in which ductular obliteration, acinar loss, and exuberant interstitial fibrosis with concentric perivascular accentuation are noted (70) (Figure 16-4). Gummas are rare.
insufficiency or even diabetes mellitus can ensue (67). Mumps is a classic cause of pancreatitis in late childhood. Diffuse necrosis and pseudocyst formation has been reported in a surgical specimen (66). Coxsackievirus may selectively involve the exocrine or endocrine components of the pancreas with parenchymal hemorrhage (65,66). Parainfluenza 3 pancreatitis, confirmed by immunohistochemistry, produced multinucleated giant cells in the pancreas of a child with severe combined immunodeficiency. Varicella-zoster infection (chicken pox) resulted in pancreatitis in individuals succumbed to the disease. The postmortem examination revealed focal pancreatic necrosis (66). Chronic coxsackievirus B interstitial pancreatitis in the absence of a meningoencephalomyocarditis in a child with α1-antitrypsin deficiency has been reported (68). Pancreatitis is a well-recognized consequence of bone marrow transplantation. Adenovirus infection may be the causative agent in some cases. In most cases of pancreatitis in children with acquired immunodeficiency syndrome (AIDS), the disease is mild and reflects systemic disease status. The incidence of opportunistic infections is low, although nonspecific inflammatory changes such as focal lymphoplasmacytic aggregates are common (69). Escherichia coli pancreatitis usually accompanies septicemia. Congenital syphilis causes a pancreatitis in which ductular obliteration, acinar loss, and exuberant interstitial fibrosis with concentric perivascular accentuation are noted (70) (Figure 16-4). Gummas are rare.
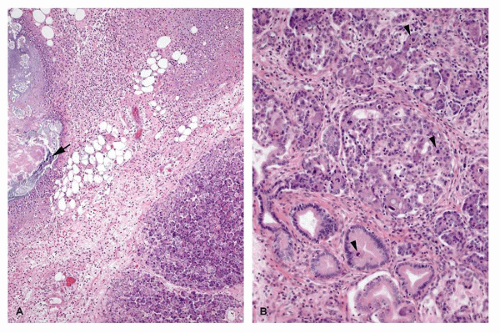 FIGURE 16-13 • Acute pancreatitis. A: Traumatic pancreatitis in a 7-year-old child showing fat necrosis with saponification (arrow) and acute inflammatory infiltrate. (Hematoxylin and eosin stain, original magnification ×100.) B: Cytomegalovirus pancreatitis with inclusions in acinar, ductal, and endocrine cells (arrow heads). (Hematoxylin and eosin stain, original magnification ×200; courtesy of James E. Dimmick, M.D., Vancouver, British Columbia.) |
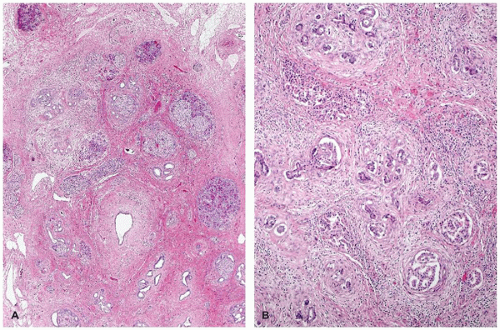 FIGURE 16-14 • Chronic pancreatitis. A: Obstructive pancreatitis with fibrosis, duct ectasia, acinar atrophy, and relative abundance of endocrine islets. Several pancreatic stones were also recovered. (Hematoxylin and eosin stain, original magnification ×50.) B: Pancreas of a 79-dayold infant (born at 30 weeks of gestation) with immunodysregulation, polyendocrinopathy, and enteropathy; X-linked inheritance syndrome (IPEX) shows diffuse mononuclear cell infiltrate, fibrosis, and acinar loss. Residual endocrine islets are present. |
Inflammatory Pancreatitis
Pancreatitis can accompany childhood collagen vascular diseases, such as lupus, but it may be difficult to distinguish between the effects of disease and those of treatment, particularly drugs. Pancreatitis has been described in Henoch-Schönlein purpura, Reye syndrome, hemolytic-uremic syndrome, and Crohn disease.
Immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is a rare X-linked recessive disorder of immune regulation manifesting with neonatal onset diabetes mellitus, severe enteropathy, eczema, anemia, thrombocytopenia, and hypothyroidism. The disorder had been known by alternative names including X-linked polyendocrinopathy, immune dysfunction and diarrhea, and X-linked autoimmunity and allergic dysregulation. A mutant mouse strain, scurfy (sf) resembles IPEX, and the disease-causing gene Foxp3 encoding scurfin was identified. Subsequently, the human IPEX locus was mapped to Xp11.23-q13.3, and mutations have been identified in the human ortholog (JM2, FOXP3) (71). Patients with protein-truncating mutations have been reported to demonstrate an absence of FOXP3-nuclear positive lymphocytes in their small and large intestines (72). Postmortem pancreatic histology is almost always abnormal; the findings include mild-to-dense lymphocytic infiltrate, acinar loss with fibrosis (chronic sclerosing pancreatitis), dilated ducts, and cystic changes (Figure 16-14). Islets of endocrine cells are decreased or absent in most cases with severe diabetes mellitus (73).
Biliary/Obstructive Pancreatitis
Some of the causes of biliary/obstructive pancreatitis in children have already been mentioned in the section on congenital malformations, such as annular pancreas, gastric duplications that connect to the pancreatic duct system, and anomalies of the pancreaticobiliary junction. Other causes include choledochal cyst, biliary sludge, and gallstone (74). Children with biliary/obstructive pancreatitis are more likely to present with jaundice and elevated serum levels of amino transferases. Cystic fibrosis is the prototype of a chronic obstructive pancreatitis.
Drug-Induced Pancreatitis
In most studies, medications attribute to less than one quarter of pediatric acute pancreatitis. The 2007 American Gastrointestinal Association Technical Bulletin on Acute Pancreatitis lists and categorizes medications as having “definite,” “probable,” or “possible” association with acute pancreatitis (75). To be included as drug-associated pancreatitis, the medication needs to be taken before the onset of acute pancreatitis (76). The most common medications in pediatric studies are valproic acid, L-asparaginase, prednisone, 6-mercaptopurine, mesalamine, and trimethoprim/sulfamethoxazole. However, interpretation of the causeand-effect relation is difficult because many patients who are on the listed medications have systemic illnesses that may predispose to pancreatitis. The comorbidities include seizure disorders, acute lymphocytic leukemia, Crohn disease, ulcerative colitis, transplants/graft versus host disease, AIDS, asthma, renal disorders, and acute myeloid leukemia (76).
Hereditary Pancreatitis
The concept of hereditary pancreatitis was first described by Comfort and Steinberg in 1952 as “chronic relapsing pancreatitis” in a family of three generations. More than 200 kindreds have since been documented, with variable prevalence worldwide. It is an autosomal dominant disorder with an 80% penetrance and variable expressivity. Classically, hereditary pancreatitis diagnosis is defined by the presence of recurrent acute pancreatitis or chronic pancreatitis occurring in two first-degree relatives or three or more second-degree relatives, in two or more generations in the absence of precipitating factors (77). The discovery of mutations within the cationic trypsinogen gene, also referred to as serine protease 1 gene (PRSS1), has resulted in the development of genetic testing. Currently, the diagnosis can also be established by the presence of a mutation of the PRSS1 gene with or without clinical or radiologic manifestations of chronic pancreatitis. Cationic trypsinogen is one of the three isoforms of the digestive proenzyme trypsinogen and represents approximately two-thirds of total trypsinogen in the pancreatic juice. Activation of trypsinogen to trypsin normally occurs in the duodenum by the brush-border localized enterokinase and also by autoactivation by trypsin. The mutations either increase stability or increase autoactivation of trypsin (77).
A major motivation for identifying patients with hereditary pancreatitis is the increased risk for developing pancreatic adenocarcinoma. Despite this increased risk of cancer, however, a national study performed in France demonstrated that hereditary pancreatitis patients do not have an excess mortality risk as compared with the general population, irrespective of gender, tobacco use, or diabetes mellitus (78).
The histopathology of PRSS1 hereditary pancreases varies among reports. An earlier study of two adult pancreatic specimens described paucicellular thick fibrosis and distorted and dilated ducts, without dysplasia or a mention of fatty replacement (79). Singhi et al. examined 12 total pancreatectomy specimens and suggested a sequential pattern of changes with increasing age (80). In pediatric patients, the pancreas was generally grossly normal, but there was microscopic variation in lobular size and shape, and parenchymal loss in the central portion of the pancreas accompanied by loose perilobular and interlobular fibrosis. The periphery was, however, replaced by mature adipose tissue. With older individuals, the pancreas showed progressive atrophy and extensive replacement by mature adipose tissue with scattered endocrine isles consistent with lipomatous atrophy. Smaller intra- and interlobular ducts were scarred, and the larger interlobular ducts were dilated with intraductal calcification. Pancreatic intraepithelial neoplasia (PanIN), a precursor of infiltrating ductal adenocarcinoma, was identified in only 3 patients. No high-grade PanINs (PanIN-3) was identified. This report is in contrast to a study by Rebours et al. (81), which identified PanIN lesions in 10 of 13 partial
pancreatectomy specimens. One 7-year-old boy showed a focus of high-grade dysplasia (PanIN-3).
pancreatectomy specimens. One 7-year-old boy showed a focus of high-grade dysplasia (PanIN-3).
Genetic Contributing Factors in Chronic Pancreatitis
Identification of genetic alterations in hereditary pancreatitis has provided a significant tool to investigate other pathogenic factors of acute and chronic pancreatitis. Chronic pancreatitis is a persistent inflammation of the pancreas that results in irreversible morphologic changes and impairment of both exocrine and endocrine functions. Genetic studies of familial and so-called idiopathic forms of chronic pancreatitis have characterized four other susceptibility genes. Witt et al. reported a close linkage of sequence alterations in SPINK1 to chronic pancreatitis. SPINK1 encodes a natural antagonist of trypsinogens, serine protease inhibitor, Kazal type I, also known as pancreatic secretory trypsin inhibitor, which binds reversibly to trypsin and inhibits its activity. Loss-offunction variants in trypsin-degrading enzyme chymotrypsin C gene (CTRC) increase the risk of chronic pancreatitis in European and Asian populations. Most recently, variants in the gene encoding carboxypeptidase A1 (CPA1) have been shown to be strongly associated with early-onset chronic pancreatitis (82). CPA1 is one of the isoforms of digestive carboxypeptidases that hydrolyzes C-terminal peptide bond. After trypsinogens, proCPA1 is the second largest component of pancreatic juice. Sequence variations of CFTR, the gene responsible for cystic fibrosis, are also associated with chronic pancreatitis. PRSS1, SPINK1, CTRC, and CPA1 are expressed in the acinar cells in the human pancreas, while CFTR is expressed in the ductal and centroacinar cells. The expressed protein CFTR is believed to dilute and alkalinize the protein-rich acinar secretions, preventing the formation of protein plugs that predispose to pancreatic injury. There is evidence to support the hypothesis that risk of pancreatitis is related to the balance between degrees of pancreatic acinar preservation and pancreatic ductal obstruction, which are associated with CFTR dysfunction in opposing directions (83). Coinheritance of a CFTR mutation that inhibits bicarbonate conductance but maintains chloride conductance and SPINK1 variants is shown to increase risk of pancreatitis. However, another report argues that the contributions of CFTR variants to chronic pancreatitis in transheterozygous status are overestimated (84).
Idiopathic chronic pancreatitis has been a traditional clinical diagnosis describing the lack of an identifiable cause. As more genes and/or more mutations are identified that cause or predispose to chronic pancreatitis, the number of patients with “idiopathic” disease seems to be decreasing. At the same time, the spectrum of diseases associated with the CTFR mutant genes keeps expanding.
Exocrine Tumors
Primary exocrine pancreatic tumors are rare in children. Due to the very small number of cases, it has been difficult to predict long-term outcome and develop and optimize therapeutic trials. As a first step, several retrospective and prospective data collections on childhood pancreatic tumors have been undertaken in the last decade in the North America (85,86), Germany (87), Italy, (88), and France (89). The primary exocrine pancreatic tumors listed in these reports are pancreatoblastoma, solid pseudopapillary neoplasm, acinar cell carcinoma, and pancreatic duct carcinoma, and these four are described below. Rare descriptive reports of mucinous cystadenoma and acinar cell cystadenoma appear in the literature (86,90). Tumors and masses that may occur, but are not specific to pancreas, include vascular lesions (26), lymphomas, and other childhood sarcomas (e.g., rhabdomyosarcoma).
Pancreatoblastoma
Pancreatoblastoma (91), also called pancreaticoblastoma, is an epithelial neoplasm that exhibits multiple lines of differentiation including acinar differentiation, often with a lesser degree of endocrine and ductal differentiation, and is associated with squamoid corpuscles (92). A distinct mesenchymal component can also be seen. Some view this as the infantile or “blastomatous” form of acinar cell carcinoma. In support of this interpretation, a considerable overlap exists among pancreatoblastoma and acinar cell carcinoma (93). Pancreatoblastomas occur most frequently in young children, but rare congenital cases discovered by prenatal imaging have been reported (94). There is a close association of infantile and congenital pancreatoblastomas with Beckwith-Wiedemann syndrome.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


