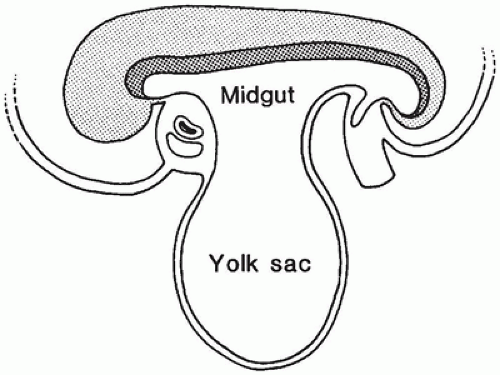
The gastrointestinal tract forms during the 4th week as the cephalocaudal and lateral folds of the trilaminar germ disk develop and incorporate the dorsal part of the endoderm-lined yolk sac into the body cavity to form a tubelike gut. During early embryonic life, the vitelline or omphalomes-enteric duct provides an open connection between the midgut and the yolk sac (Figure 14-1). This connection becomes progressively longer and narrower as gestation proceeds and eventually forms part of the umbilical cord. By week 10, the communication between the lumen of the midgut and the umbilicus becomes obliterated and soon disappears.
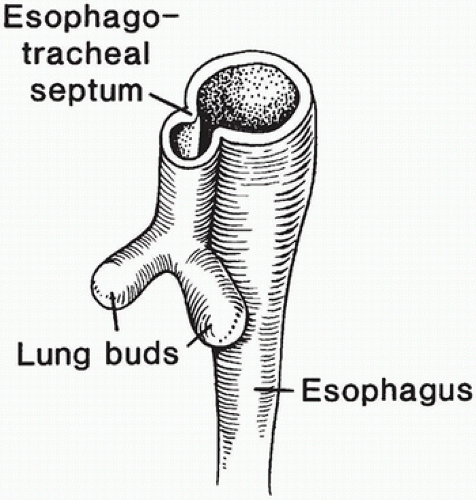
The laryngotracheal diverticulum develops from the ventral foregut during week 4 of gestation (Chapter 12). Gradual formation of an esophagotracheal septum along the length of the laryngotracheal diverticulum separates the ventral respiratory and the dorsal digestive tubes (Figure 14-2). The common origin of the trachea and esophagus from the foregut results in various forms of fistulas if separation is incomplete.
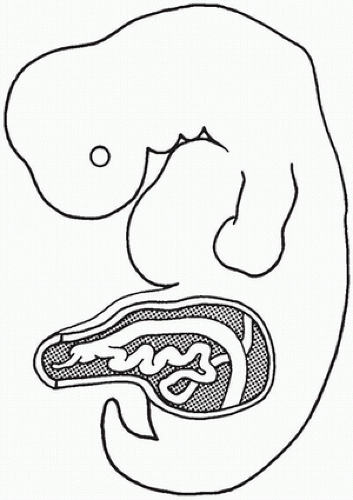
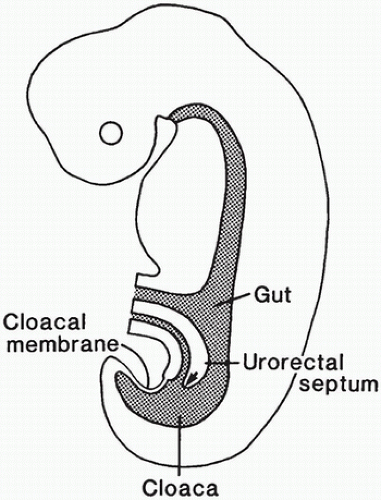
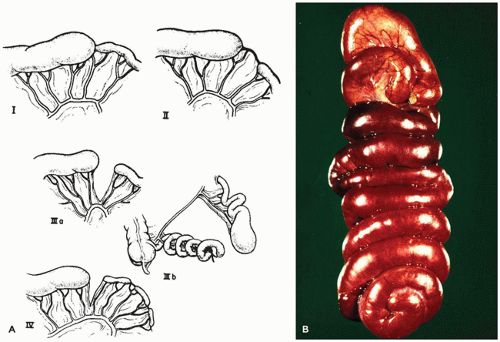
During the 2nd month of embryonic life, rapid cellular proliferation within the digestive tube results in a transient partial obliteration of the duodenal lumen, the so-called solid stage of development. Recanalization occurs by week 8 of gestation. Rapid midgut growth within the relatively small body cavity results in a temporary herniation of the lengthening midgut into the umbilical stalk during weeks 6 to 11 (Figure 14-3). During this physiologic herniation, the intestinal loops rotate counterclockwise, a process that continues as the intestinal loops return to the abdominal cavity during weeks 10 and 11, so that the cecum comes to lie in the right side of the abdomen. If this orderly process fails to occur or is anomalous, the locations of the small and large intestine, mesentery, and fixation points of the intestine to the body wall will be abnormal. The hindgut, or posterior portion of the primitive digestive tube, initially ends posteriorly in the cloaca, separated from superficial ectoderm by the cloacal membrane (Figure 14-4). A transverse ridge, the urorectal septum, grows posteriorly from the umbilical stalk and gradually divides the cloaca into a ventral portion, the urogenital sinus, and a dorsal portion, the future rectum and anus. This division is normally complete at the end of week 6 of gestation. The membrane covering the anal canal disappears by week 9, so that communication between the digestive tract and the amniotic cavity is established caudally.
DISORDERS OF THE ESOPHAGUS
Congenital Abnormalities
Persistent Embryonic Epithelium
The embryonic and early fetal esophagus is lined by ciliated stratified columnar epithelium. The transformation to stratified squamous epithelium is usually complete by week 25 of gestation, but occasionally, a patch of superficial columnar epithelium persists at birth, especially in premature infants. Persistent embryonic epithelium is usually found incidentally at autopsy as microscopic foci in either the proximal or distal end of the esophagus and is of little clinical consequence. This change is limited to surface epithelium only; glandular mucosa, as in gastric heterotopia of the esophagus, is not present. Because it is not usually found after early infancy, persistent embryonic epithelium is presumed to be replaced by squamous epithelium.
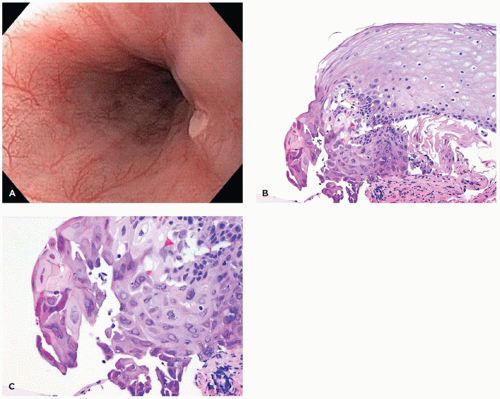
Heterotopic Gastric Mucosa (Inlet Patch)
Single or multiple small patches (5 to 30 mm) of gastric cardiac- or fundic-type mucosa can sometimes be found incidentally in the cervical esophagus (eFigure 14-1). The incidence in patients undergoing esophagogastroduodenoscopy has been reported to be up to 10% (1). These patches of heterotopic gastric epithelium usually are not clinically important and are rarely biopsied since most endoscopists are familiar with them. In contrast to Barrett esophagus (BE), inlet patches occur in the upper esophagus and are surrounded by squamous mucosa; however, confusion with BE can occur if the endoscopic findings are not communicated to the surgical pathologist. They can be colonized by Helicobacter pylori organisms (1).
FIGURE 14-1 • During week 4 of gestation, head and tail folds of the embryo surround portions of the yolk sac. An open connection between the primitive midgut and the yolk sac exists. After this connection narrows, it is known as the vitelline or omphalomesenteric duct.
Esophageal Duplication
GI duplications represent an array of lesions with a confusing terminology that includes enteric remnants, dorsal enteric cysts, neurenteric cysts, and posterior mediastinal cysts. The simple classification proposed by Dimmick and Hardwick (2), which divides these lesions into duplications and neurenteric (dorsal enteric) cysts, is preferred. Duplication of the esophagus is rare. The duplicated segment may be a separate cylindrical tube alongside part of the normal esophagus with a complete mucosa, submucosa, and two-layered muscularis externa (double esophagus). Alternatively, a spherical, intramural esophageal cyst may form and share a portion of muscularis propria with the adjacent esophageal wall. Esophageal duplication occurs most often in the thorax adjacent to the distal two-thirds of the esophagus, but it may also occur in the lateral cervical area. Esophageal duplication cysts may be asymptomatic and discovered incidentally, or they may cause tracheal or esophageal compression. The epithelium is either stratified squamous or columnar; the latter is derived from persistent embryonic esophageal ciliated columnar epithelium. Distinction between esophageal and bronchogenic cysts may be difficult because they occur at similar locations in the mediastinum and show similar ciliated columnar epithelium. The diagnosis of esophageal cyst is made if a twolayered muscularis externa is present. A bronchial origin is favored if cartilage or respiratory glands are identified. The generic designation of “foregut cyst” is used in cases in which the lining epithelium is primitive columnar without the distinguishing features cited.
FIGURE 14-2 • Development of the respiratory system from the foregut at week 4 of gestation. The esophagotracheal septum develops from the two lateral folds that migrate toward the midline to separate the developing respiratory diverticulum from the primitive gut.
FIGURE 14-3 • Physiologic gut herniation in an embryo at week 8 of gestation. Rapid elongation of the intestine in a relatively small abdominal cavity causes the gut to herniate into the umbilical cord. This herniation resolves at the end of the 3rd month of gestation. A failure in the normal events at this stage explains the omphalocele and malrotation.
FIGURE 14-4 • Primitive hindgut region in an embryo at 6 weeks of gestation. The urorectal septum grows posteriorly to divide the cloaca into a urogenital portion separate from the intestinal portion. Note the intact cloacal membrane.
Neurenteric Cyst (Remnant) of the Mediastinum
Neurenteric remnants include diverticula, fistulas, cysts, and fibrous cords, which originate from the dorsal midline of the gastrointestinal tract and attach to or pass through the vertebral column and spinal cord cranial to their enteric origins and may continue to the skin of the dorsal midline overlying the involved vertebra. They are located at any level but are most common in the cervicothoracic and lumbosacral area. When located in the posterior mediastinum, neurenteric cysts may be confused with esophageal duplication cysts. Most are associated with vertebral anomalies, and about 25% have intraspinal anomalies (3). The walls of these remnants or cysts can be composed of all the normal layers of the gastrointestinal tract, and neuroglial tissue is found in the lesions involving the spinal cord and vertebrae. The epithelial lining is most often gastric mucosa, which can result in ulceration, perforation, and hemorrhage. Small-intestinal mucosa and primitive columnar epithelial lining have also been described.
Esophageal Atresia and Tracheoesophageal Fistula
Esophageal atresia and tracheoesophageal fistula occur together in most cases. The dual anomalies, which occur in approximately one in 3000 births, result from faulty division of the foregut into tracheal and esophageal channels during the first month of embryonic life. Additional congenital anomalies (usually midline) occur in 50% of these infants, directly affecting the prognosis. Congenital heart diseases, especially ventricular septal defect, patent ductus arteriosus, and tetralogy of Fallot, are seen in 30% of cases of esophageal atresia, and imperforate anus occurs in approximately 10%. In babies with multiple malformations, the VATER (vertebrae, anal, tracheoesophageal, radial, and renal anomalies) association or the VACTERL (vertebrae, anal, cardiac, tracheoesophageal, renal, and limb) association should be considered. Approximately one-third of the infants with esophageal atresia are born prematurely, so that morbidity is further increased.
Variations in the anatomy of esophageal atresia and tracheoesophageal fistula are diagrammed in Figure 12-10 (see Chapter 12). Esophageal atresia without tracheoesophageal fistula occurs rarely, and tracheoesophageal fistula without esophageal atresia (H-type fistula) is even more unusual. The most common type is esophageal atresia with distal tracheoesophageal fistula, which accounts for 85% of the cases (Figure 14-5). The esophagus ends in a blind pouch in the upper chest, and the lower portion of the esophagus is connected to the trachea at or near the carina by a tracheoesophageal fistula less than 0.5 cm in diameter (see Figure 12-10). During breathing, air enters the stomach through the fistula, and as the stomach becomes distended, gastric secretions pass through the fistula into the lungs, causing pneumonia. The infant cannot swallow oral secretions or food; attempts at feeding produce regurgitation and aspiration. The diagnosis is suggested by the inability to pass a tube from the mouth to the stomach and is confirmed by plain x-ray films of the chest and abdomen. Treatment is surgical and consists of extrapleural transection of the fistula and anastomosis of the two ends of the esophagus.
FIGURE 14-5 • Tracheoesophageal fistula at autopsy (posterior view of thoracic organ block). The probe is in the blind upper portion of the esophagus, and the tracheoesophageal fistula arises at the tracheal bifurcation.
Isolated esophageal atresia without an associated fistula, found in 7% to 8% of cases, is usually characterized by blind proximal and distal esophageal pouches, often separated by a wide gap. Multiple surgical procedures are usually required for repair.
In the relatively rare cases of isolated tracheoesophageal fistula without esophageal atresia (H-type fistula), the diagnosis is often delayed beyond the newborn period. Patients with this anomaly present with coughing or choking during feeding and with recurrent pneumonia. Histologic study of the tracheoesophageal fistula often reveals foci of primitive ciliated columnar epithelium, respiratory glands, and even cartilage. These tracheobronchial elements and abnormalities in smooth muscle may extend for some distance into the distal esophagus (tracheobronchial remnant-choristoma).
Esophageal Stenosis
In most cases, esophageal stenosis is an acquired lesion caused by gastroesophageal reflux (GER) with severe peptic esophagitis. However, rare forms of congenital esophageal stenosis have been described, resulting from tracheobronchial remnants, fibromuscular hypertrophy, or membranous mucosal rings and webs (4). About half of the cases are associated with atresia. Sloughing of esophageal mucosa occurs in inherited epidermolysis bullosa, which is complicated by stenosis.
Acquired Diseases
Esophagitis Due to Gastroesophageal Reflux
GER is common during the first few months of life, as evidenced by the frequent occurrence of effortless regurgitation at this age. It is considered a physiologic process secondary to immature esophageal peristaltic and lower esophageal sphincter function and gradually improves during the first year of life. Physiologic reflux is a normal function that serves a protective role in the postprandial period (5). GER disease (GERD) refers to reflux associated with mucosal damage or with symptoms impairing quality of life. Reflux esophagitis is reported to occur in 2% to 5% of the population, and esophagitis is present in 15% to 60% of children with symptoms of reflux (5).
The symptoms of reflux esophagitis differ with the age of the patient. Infants show effortless regurgitation and sometimes forceful vomiting, excessive irritability, apnea, and failure to thrive as a consequence of caloric losses. Older children present with vomiting and poorly characterized abdominal or chest pain. Patients of any age may exhibit gastrointestinal blood loss (from esophageal ulceration), failure to thrive, and recurrent pulmonary problems (e.g., asthma, pneumonia, and night cough). Most children with reflux esophagitis are otherwise normal, but certain groups of children are predisposed, including those with neurodevelopmental problems, cystic fibrosis, and bronchopulmonary dysplasia and those who have undergone repair of esophageal atresia and tracheoesophageal fistula in infancy. Esophageal pH monitoring, esophageal manometry, and barium esophagography may be used in patient evaluation. If a patient has signs of esophagitis, such as pain, gastrointestinal blood loss, or failure to thrive, esophagoscopy and esophageal biopsy are indicated.
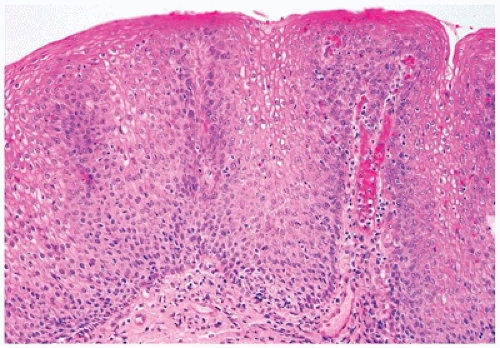
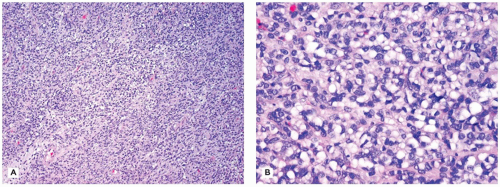
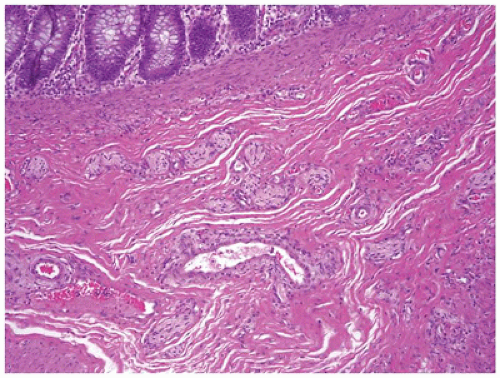
Histologic features in children with reflux esophagitis are similar to those widely described in adults with the same condition (Figure 14-6). Diagnostic histologic findings include intraepithelial inflammation with eosinophils, lymphocytes, and neutrophils and epithelial changes such as basal cell hyperplasia (>15% to 20% of total epithelial thickness), papillary elongation (>50% of epithelial thickness), and spongiosis. These findings may be noted even in endoscopically normal mucosa (6). Epithelial changes correlate well with pH monitoring studies and may be the only histologic features present when patients have begun treatment with antireflux agents. However, basal cell hyperplasia and papillary elongation may be difficult to assess in poorly oriented biopsies. They are also common in biopsies obtained from near the squamocolumnar junction (Z-line) in patients without reflux. Spongiosis, also a sensitive marker of epithelial cell injury, is usually best observed in the lower cell layers and can be evaluated even in poorly oriented biopsies. Intraepithelial eosinophils appear to be the earliest and most specific correlate of reflux esophagitis in infants. It should be noted that eosinophils can also be present in other conditions such as Crohn disease (CD), food impaction and infections and are particularly prominent in eosinophilic esophagitis (EoE). Lymphocytes and Langerhans cells are normally present in the esophageal squamous mucosa, so the determination of an abnormal increase in these cells is subjective. Increased intraepithelial lymphocytes (IELs) may be noted in chronic inflammatory bowel disease (IBD), in celiac disease, and in association with H. pylori gastritis. In a recent study in the pediatric population, a marked increase in IELs (>50/hpf) was associated with CD and found in 28% of patients with CD (7).
FIGURE 14-6 • Reflux esophagitis. Note the presence of basal cell hyperplasia and lengthening of the papillae. There are also scattered intraepithelial eosinophils (hematoxylin and eosin, 200×).
In otherwise healthy pediatric outpatients, occasional cases of infectious esophagitis, particularly herpes simplex esophagitis, present with signs and symptoms mimicking those of reflux esophagitis. Ingestion of caustic substances, CD, and dermatologic conditions, such as bullous pemphigoid and Stevens-Johnson syndrome, are rare possibilities, and other suggestive clinical findings are usually present. In children who are immunosuppressed or severely debilitated from another illness, infectious esophagitis is an important diagnostic consideration (Table 14-1).
The treatment of GERD consists primarily of acid suppression and enhancing gastric emptying. Acid blockade with antacids, H2 receptor antagonists, or proton pump inhibitors are the mainstay of GERD therapy. Gastroesophageal fundoplication is performed in cases refractory to medical therapy or strictures or those at risk for pulmonary complications; failure rates of 5% to 20% have been reported (5). Sequelae of GER include ulcers (usually of the distal one-third of the esophagus and often associated with blood loss), stricture, and BE.
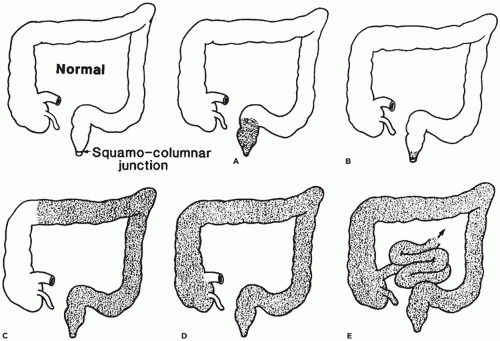
Barrett Esophagus
BE, in which columnar epithelium replaces the normal squamous lining of the distal esophagus, is an acquired metaplastic condition caused by chronic GER. It is now established that even in children, BE is invariably found in association with severe reflux esophagitis; however, it is rare in children, occurring in only a small percentage that undergo biopsy for symptomatic GER. Usually, older children, not infants, are affected. BE cannot be predicted by the clinical presentation; the symptoms are those of the associated reflux esophagitis.
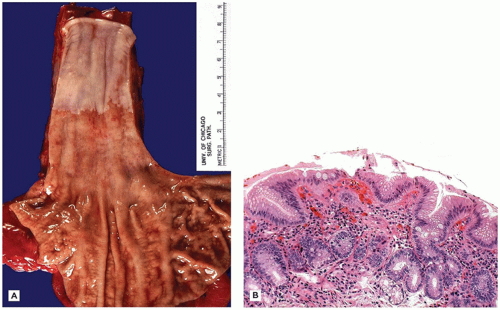
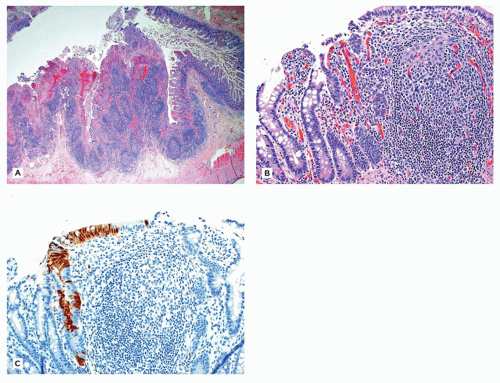
The changes in BE affect the lower portion of the esophagus (Figure 14-7A) and involvement may be either circumferential or patchy. BE is currently defined by the American College of Gastroenterology as endoscopically recognizable columnar metaplasia of the normal esophageal squamous mucosa that is histologically confirmed to have intestinal metaplasia, defined by the presence of goblet cells (Figure 14-7B) (8). The squamous mucosa proximal to the affected esophagus often shows changes of reflux esophagitis. Well-formed barrel-shaped goblet cells are usually easily recognizable with ordinary hematoxylin and eosin staining, but recognition can be enhanced and confirmed by staining with Alcian blue at pH 2.5, which imparts a blue color to intestinal-type acidic mucins. A pitfall is that Alcian blue-positive cells can be observed in the gastroesophageal junction of normal fetuses and young children, in the absence of goblet cells. In Great Britain, columnar-type mucosa without goblet cells is accepted as diagnostic of BE, provided the endoscopist is certain that the biopsies were obtained from the tubular esophagus and not the proximal stomach (9). Thus, the location of the biopsy site in relation to the lower esophageal sphincter must be known by the pathologist before BE can be diagnosed. However, because the endoscopic landmarks used to separate esophagus and stomach (primarily the upper extent of the gastric rugal folds) are not precise, particularly in the presence of a hiatal hernia, confusion between distal esophagus and gastric cardia is possible. Barrett mucosa has the potential to progress to dysplasia and, after many years, to adenocarcinoma in about 1% to 2% of cases. Patients with BE need to undergo routine, frequent monitoring, although there currently is no consensus on the frequency of surveillance in children. Adenocarcinoma in BE in children is very rare but has been reported in a patient as young as 8 years old (10).
TABLE 14-1 CAUSES OF ESOPHAGITIS IN CHILDREN
Gastroesophageal reflux
Allergy
Food impaction
Infections
Ingestion of corrosive agents
Prolonged retention of medication pills
Trauma
Repaired esophageal atresia
Crohn disease
Radiation
Motility disorder
Eosinophilic Esophagitis
EoE is a relatively recently described condition in which patients with a severe esophageal eosinophilia present with symptoms that are otherwise indistinguishable from those secondary to reflux esophagitis but fail to respond to conventional antireflux therapy or antireflux surgery. A recent set of diagnostic guidelines were developed for EoE in 2011 and include the following: (a) patients present with an isolated esophageal eosinophilia (other GI histology is normal) and symptoms of esophageal dysfunction; (b) the degree of eosinophilia in these patients is almost always greater than 15 eosinophils per high-magnification microscopic field; (c) disorders such as GERD and PPI-responsive esophageal eosinophilia need to be excluded; and (d) the symptoms and histologic abnormalities improve with either steroid or restriction diet therapy (11). The frequent association of extraintestinal allergic symptoms (asthma, eczema, and chronic rhinitis) in these patients has led to the hypothesis of an antigen- (food)/immune-mediated etiology for this type of esophagitis. It is still a matter of debate whether a damaged mucosa, say by reflux, could predispose to the development of EoE or whether changes in food processing are to blame. In young children, presenting symptoms include difficulty feeding, prolonged irritability and crying, failure to thrive, and growth delay. Adolescents and adults diagnosed with this disorder often suffer from significant dysphagia and the development of esophageal strictures. Esophageal fibrosis in the deeper submucosal and muscular layers occurs in many patients with EoE, and left untreated, EoE may cause a severe narrowing of the esophagus.
FIGURE 14-7 • Barrett esophagus. A: Esophagectomy specimen exhibiting a 5-cm circumferential segment of Barrett mucosa. B: Barrett mucosa, characterized by specialized columnar mucosa with goblet cells (hematoxylin and eosin, 200×).
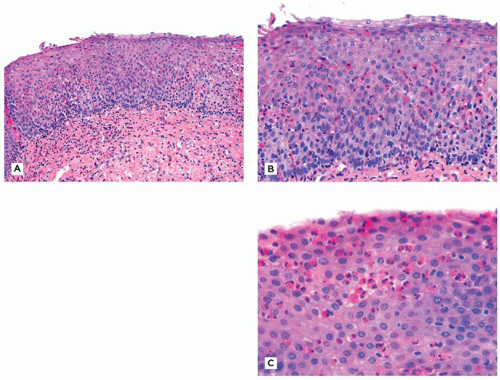
Characteristic endoscopic findings include wrinkled or thickened esophageal squamous mucosa, sometimes with circumferential rings, linear furrows, or tiny vesicles. Endoscopic biopsies of the esophagus typically reveal a heavy but sometimes patchy infiltrate of eosinophils, frequently forming microabscesses near the luminal surface (Figure 14-8A to C). As previously mentioned, 15 eosinophils in any single high-power field (HPF) (400×) should be regarded as consistent with EoE in the proper clinical context (12). Other features include extracellular eosinophil granules, basal cell hyperplasia, and lamina propria fibrosis.
The presence of inflammatory changes that are equally severe in biopsies from the mid esophagus and distal esophagus is also a useful finding in making the diagnosis of EoE (since reflux changes are typically more severe distally than proximally). The presence of admixed neutrophils, on the other hand, favors the presence of reflux esophagitis, since in general, only eosinophils are present in EoE unless ulceration has occurred. Biopsies of the gastric cardia can also be useful in distinguishing between reflux esophagitis and EoE. In reflux esophagitis, the cardia is uniformly inflamed (i.e., “carditis”), while in EoE, the cardia is typically not inflamed. The presence or absence of increased eosinophils in any gastric or duodenal biopsies obtained during the endoscopy should also be mentioned in the surgical pathology report, to address the possibility of a more generalized eosinophilic gastrointestinal disorder. In some cases, it is not possible to make a firm histologic distinction between EoE and severe reflux esophagitis. Correlation with the clinical history and the endoscopic appearance is often sufficient to arrive at the proper diagnosis, but 24-hour esophageal pH monitoring may be necessary in some patients. Other causes of esophageal eosinophilia need also be considered (Table 14-2).
The current treatment for EoE includes topical swallowed corticosteroids or systemic steroids, hypoallergenic diets, or combination of each. Esophageal dilatation may be required for strictures that are unresponsive to medical therapy.
Infectious Esophagitis
Infectious esophagitis is rare except in hospitalized, immunosuppressed, and debilitated children, who are at significant risk for the development of esophagitis in association with infection by Candida species, herpes simplex virus, and cytomegalovirus (CMV). Bacterial infection is a practical consideration only as a superinfection.
FIGURE 14-8 • Eosinophilic esophagitis. A: Low power showing basal cell hyperplasia. Note the fibrosis of the subepithelial stroma. B: There are more than 40 eosinophils per high-power field. C: An eosinophilic microabscess in the superficial epithelium (hematoxylin and eosin, A: 100×, B: 200×, C: 400×).
Herpes Simplex Esophagitis
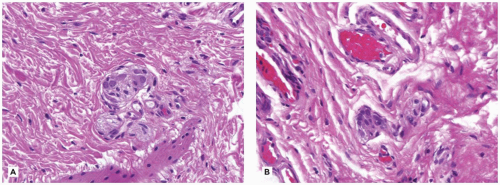
Herpes esophagitis presents as odynophagia and is often accompanied by gingivostomatitis. Multiple small, discrete ulcers separated by normal mucosa are distributed throughout the esophagus (Figure 14-9A). In severe cases, confluent ulceration can occur. Microscopically, severe necrotizing esophagitis, abundant neutrophils, and ulceration are found. Epithelial cells at ulcer margins often demonstrate discrete eosinophilic intranuclear inclusions (Cowdry type A) or ground-glass intranuclear inclusions (Cowdry type B) (Figure 14-9B, C). Only the squamous cell can be infected by the herpes simplex virus; so if the biopsy consists only of granulation tissue and necrotic debris, no comment can be made regarding the presence or absence of this infection but IHC for HSV1 or HSV2 may be helpful. Although it occurs most often in immunosuppressed persons, herpetic esophagitis may occasionally be found in otherwise normal children.
TABLE 14-2 CAUSES OF ESOPHAGEAL EOSINOPHILIA
Eosinophilic esophagitis
Gastroesophageal reflux disease
PPI-responsive esophageal eosinophilia
Eosinophilic gastroenteritis
Crohn disease
Hypereosinophilic syndrome
Achalasia
Vasculitis, pemphigus, connective tissue disease
Infection
Food impaction
Candida Esophagitis
Immunosuppression, premature birth, cancer chemotherapy, and AIDS are the most significant risk factors for the development of esophageal candidiasis in infants and children, although infection can also occur in patients without predisposing illnesses (13). Esophageal involvement is common in children with mucocutaneous candidiasis. In debilitated patients, esophageal infection may lead to systemic candidiasis. The gross appearance is usually a combination of white plaques and ulcerations. Histologically, the plaques consist of masses of pseudohyphae and yeast forms admixed with inflammatory debris and fibrin (eFigure 14-2A to C). Superficial collections of neutrophils should alert to the presence of organisms.
Cytomegalovirus Esophagitis
CMV esophagitis is uncommon and limited to immunosuppressed persons. It rarely occurs alone; it is usually part of a systemic CMV infection or an infection involving the whole gastrointestinal tract. In contrast to herpes simplex esophagitis, CMV cannot infect squamous epithelial cells, and the viral cytopathic effect is seen in stromal elements, endothelium, and submucosal glandular epithelium; so if the biopsy consists only of squamous epithelium, no comment can be made regarding the presence or absence of this infection.
FIGURE 14-9 • Herpes simplex virus esophagitis. A: Endoscopic appearance of a midesophageal ulcer. B: Viral inclusions in squamous epithelium at the edge of the ulcer 200×. C: High power to demonstrate typical intranuclear inclusions and multinucleated cells 400×.
DISORDERS OF THE STOMACH
Congenital Anomalies
Hypertrophic Pyloric Stenosis
Although pyloric stenosis (PS) most likely presents between the 2nd and 4th week of life, there have been reports of PS in the newborn as well as in infants aged 4 months (14). PS is a common condition, seen in 1 of 200 infant boys. The male-to-female ratio is 5:1 or greater, and white, firstborn boys are at greatest risk. An increased concordance rate in twins and increased recurrence rate in siblings have been reported (15). Neurons supplying the circular muscle layer of the pylorus lack activity of the enzyme nitric oxide synthase. The circular muscle layer undergoes hypertrophy and elongation, and gastric outlet obstruction ensues. Progressive nonbilious vomiting, the primary manifestation, commences at 2 to 6 weeks of age in an otherwise healthy infant. The diagnosis is suggested when the hypertrophic pyloric muscle mass, approximately the size of an olive, is palpated in the right upper quadrant after a feeding. Abdominal x-ray films show marked gaseous distension of the stomach, and barium studies demonstrate a narrow and elongated pyloric channel (“string sign”). Treatment is surgical. At operation, the hypertrophic pyloric muscle appears as an elongated sphere (“olive”), approximately 2.5 cm long and 1.5 cm in diameter. A longitudinal surgical incision of the hypertrophic muscle down to the submucosa (pyloromyotomy) immediately and efficaciously relieves the obstruction. Occasional postmortem observations indicate that the circular layer of muscularis propria is hypertrophic, hyperplastic, and disorganized in appearance. The outer, longitudinal muscle layer is attenuated and of variable thickness.
Antral Web
A very unusual cause of gastric outlet obstruction in young infants is an antral web (antral diaphragm) of fibrous tissue and gastric mucosa obstructing the antrum a few centimeters proximal to the pylorus. A small, central aperture, usually no more than several millimeters in diameter, permits passage of some stomach contents; variability in the size of the opening explains the variability in age at presentation. The diagnosis is made by barium studies, and endoscopy is often difficult.
Duplication
Gastric duplication presents as a cystic mass on the greater curvature or at the pylorus and may present with bleeding, rupture, or obstruction. The pathologic features are similar to those of the more commonly encountered small-intestinal duplication. The mucosa of a gastric duplication resembles stomach mucosa in most cases, but primitive or simplified gut epithelium or intestinal mucosa is also encountered.
Pancreatic Heterotopia and Pancreatic Acinar Metaplasia
Heterotopic pancreatic tissue is most commonly noted in the stomach and proximal small bowel, although it can be seen in the liver, spleen, umbilicus, and other sites. Heterotopic pancreatic tissue also affects small-bowel stenoses, duplications, and diverticula. It is often detected incidentally on imaging studies or at autopsy. The majority of patients are asymptomatic. It usually forms a sessile mass in the antrum; a central depression may be seen, corresponding to the opening of the pancreatic duct draining the heterotopic tissue. It consists histologically of acinar and endocrine pancreas. The term adenomyoma has been applied to a variant characterized by a predominance of pancreatic duct structures interlaced with smooth muscle bundles but without pancreatic parenchyma. Occasionally, ulceration develops in the overlying mucosa and causes epigastric pain. More commonly in our experience, is the usually incidental histologic finding of clusters or small lobules of pancreatic acinar cells located in the deep gastric antral mucosa, which has been called pancreatic acinar metaplasia. This lesion is distinct from pancreatic heterotopia in that it is a histologic finding without an associated mass or recognizable endoscopic features and does not include pancreatic ducts or endocrine tissue. It has been associated with chronic gastritis in adults, whereas there are no distinct clinical or endoscopic features in children (16).
Acquired Diseases
Spontaneous Gastric Perforation in the Neonate
Spontaneous perforation of the body of the stomach is a rare but frequently fatal condition that can occur in both preterm and term infants, especially those under intensive care (17). The cause of the perforation is inapparent, although it often occurs in an area of hemorrhagic or coagulative necrosis and may be ischemic or traumatic in origin. The usual presentation is sudden abdominal distension and pneumoperitoneum.
Gastritis
A list of the types of gastritis in children is much shorter than a similar list in adults because of the absence of many of the atrophic, metaplastic, and dysplastic conditions of the adult stomach. However, it is clear that gastritis occurs with considerable frequency in children and adolescents. Numerous classification schemes exist, but for practical purposes, gastritis is categorized by etiology if apparent.
Hemorrhagic and Erosive Gastritis
The etiology of acute hemorrhagic gastritis is multifactorial, with ischemia, stress, and drug therapy playing contributory roles. Drugs known to damage the gastric mucosa include aspirin, corticosteroids, alcohol, and nonsteroidal anti-inflammatory drugs (NSAIDs), such as indomethacin. The ingestion of corrosive substances also causes a similar picture. At endoscopy, a diffusely injected and edematous mucosa, often with petechial hemorrhages and small erosions, is seen. In severe cases, which usually occur in very ill children hospitalized for sepsis, hemorrhagic shock, major surgery, burns, central nervous system disorders, or other severe illness, the changes are most severe in the gastric body and fundus.
Biopsies are usually not obtained in these severely ill patients, and therefore, this condition is usually seen at the time of autopsy. The histologic changes essentially represent a chemical injury to the gastric mucosa caused by reduced host defense against the injurious action of gastric acid and digestive enzymes. Hemorrhage and mucosal edema dominate the histologic picture. Significant inflammation is not present except directly adjacent to areas of ulceration (eFigure 14-3A, B).
Helicobacter pylori Gastritis
Since the early 1980s, it has been recognized that diffuse antral gastritis is caused by infection with H. pylori, a small coccoid or spirillar gram-negative bacillus. This organism, which is the pathogen responsible for the associated symptoms and pathologic changes, is not an opportunist or a commensal. Children with H. pylori infection usually present with nausea, vomiting, and epigastric pain. Endoscopy shows erythema, particularly in the antrum, and, in the more severe cases, erosion, antral nodularity, and thickened gastric folds. However, there is no good correlation between the endoscopic and histologic findings of gastritis. That is, in many cases where endoscopic findings of gastric mucosal erythema, granularity, or erosion are described, biopsies are entirely unremarkable. Conversely, in many cases where the gastric mucosa is described as endoscopically normal, gastritis may be evident histologically.
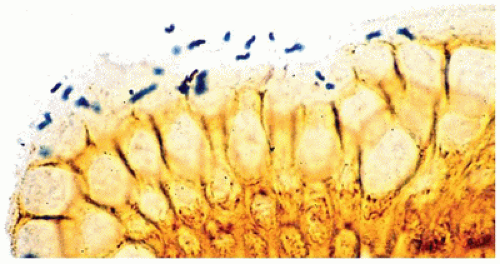
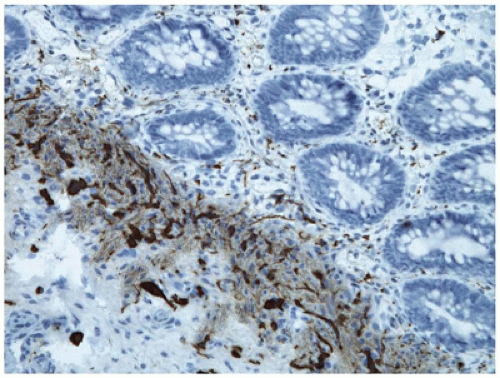
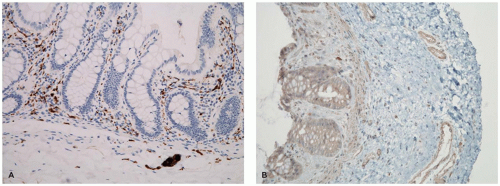
According to guidelines recently published by the North American and the European Societies for Pediatric Gastroenterology Hepatology and Nutrition (NASPGHAN and ESPGHAN), the initial diagnosis of H. pylori infection should be based on endoscopy with gastric biopsies (one each from the antrum and corpus) plus either a positive rapid urease test or a positive culture (18). The rapid urease or CLO (for Campylobacter-like organism) test is inexpensive and sensitive, and is based on the production of urease by the organism, which causes a change in the color of the test solution. Serologic studies are also sensitive, but positivity may persist for some time after eradication of the organism. On biopsy, the organisms are most reliably found in the antrum, although the fundus and cardia of the stomach may also be affected. The bacilli can be seen faintly on ordinary preparations stained with hematoxylin and eosin, but they are more easily seen with Giemsa, Genta, Warthin-Starry, or immunoperoxidase staining; they appear as small curved or slightly twisted rods, 4 to 5 µm in length, within the mucous coat overlying the surface or superficial foveolar epithelium (Figure 14-10). The presence of the organisms is almost invariably accompanied by neutrophils.
The antral mucosa exhibits a diffuse superficial infiltrate composed primarily of plasma cells and lymphocytes. Active foci of neutrophilic infiltration may be seen in the lamina propria or in glandular or surface epithelium. Although lymphoid aggregates are normal in the gastric mucosa, the presence of lymphoid follicles with germinal centers is highly suggestive of past or current H. pylori infection. In patients on a proton pump inhibitor for dyspepsia or symptoms of GER, the H. pylori organism may migrate to cause active gastritis of the gastric body mucosa, resulting in an inactive appearance of the antral gastritis.
Eradication of the organism is recommended for children with H. pylori-positive peptic ulcer disease and is based on the use of a proton pump inhibitor in combination with two antibiotics for 7 to 14 days (18). This typically results in prompt disappearance of the organisms and the neutrophilic component of the mucosal inflammatory cell infiltrates. By contrast, it may take many months for the lymphocytic and plasma cell infiltrates to disappear. Biopsies obtained during this period may be diagnosed as inactive gastritis. The diagnosis of inactive gastritis can be difficult, as there are a number of lamina propria lymphocytes and plasma cells in the gastric mucosa normally (19). As a general rule of thumb, when the density of plasma cells is such that they are clustered and touching each other, this can be regarded as indicative of inactive gastritis. In some patients with inactive antral gastritis, there may not be an antecedent diagnosis of H. pylori gastritis, as the infection may have been treated incidentally during antibiotic treatment of infection elsewhere (e.g., otitis media). Treatment failure is not uncommon and is usually related to antibiotic resistance, and options for these children usually include repeat EGD with culture and antibiotic susceptibility testing (18).
FIGURE 14-11 • Eosinophilic gastritis. A: A pure infiltrate of abundant eosinophils 200×. B: Eosinophils infiltrate the surface epithelium 400×.
Even though H. pylori causes duodenal ulcers, the organism is not found in duodenal mucosa except in instances of gastric metaplasia of the duodenum, which is rare in children. The mechanism of duodenal ulcer formation in H. pylori infection is thought to involve increased acid secretion as a response to the gastric infection, as well as direct damage by the organism in the areas of duodenal gastric foveolar metaplasia.
In addition to the immediate morbidity of gastritis and ulcer disease in children and adults, infection with H. pylori is known to carry a risk for future adenocarcinoma of the stomach and gastric lymphoma arising in mucosa-associated lymphoid tissue (MALT).
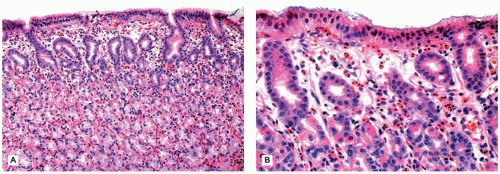
The histologic differential diagnosis of H. pylori gastritis includes a small number of unusual conditions of the stomach with distinctive clinical and histologic findings, including involvement by eosinophilic gastroenteritis (EG) (Figure 14-11), CD, chronic granulomatous disease (CGD), and Henoch-Schönlein purpura (HSP). Lymphocytic gastritis, defined as greater than 25 lymphocytes for every 100 epithelial cells in the gastric mucosa (20), is most frequently associated with celiac disease in children (eFigure 14-4). It is not likely to be associated with H. pylori infection unless neutrophils are also present (21).
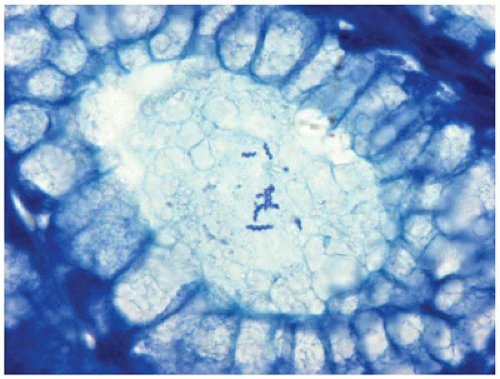
Helicobacter heilmannii infection of the stomach is more rare and not as serious or chronic a disease as is H. pylori gastritis. The clinical presentation and histologic features are similar except that H. heilmannii gastritis is more focal and less intense compared to H. pylori gastritis (22). In addition, H. heilmannii is a larger organism than H. pylori, more obviously spiraled, and more readily seen on sections stained with hematoxylin and eosin (Figure 14-12). Helicobacter heilmannii and H. pylori may coexist; the treatment is similar.
Peptic ulcers are of two types: acute (stress) and chronic. They are also classified as primary and secondary, the latter associated with systemic disease. Nearly all peptic ulcers occur in the stomach and duodenum, but they may occur in any location where acid- and pepsin-secreting gastric mucosa is found, including Meckel diverticulum.
Most cases of childhood and adult chronic ulcers have been shown to be caused by infection with H. pylori, though it has been more recently recognized that a significant proportion of chronic duodenal ulcers (20% to 40%) are not related to H. pylori, drugs, or any other identifiable cause (23). Chronic (or primary) peptic ulceration in children is the same acid peptic disease that is so common in adults. This condition can develop in children as young as 4 or 5 years old, although it is more common in preadolescents and adolescents of either sex. It is most common in adolescent boys. Duodenal ulcer is more common than is gastric ulcer. Chronic abdominal pain is the most frequent presenting symptom. More than 50% of the patients have hematemesis, melena, or occult bleeding at the time of presentation. At endoscopy, chronic peptic ulcers are usually round to oval, less than 2 cm in diameter, well delineated from the surrounding mucosa by sharp margins, and covered by exudate at the base.
Microscopically, granulation tissue and scar tissue form the ulcer base, which often extends deep into the muscularis propria. The stomach invariably shows active antral gastritis, and H. pylori is usually identified. If the ulcer is duodenal, active duodenitis is usually present in surrounding, nonulcerated mucosa. Chronic peptic ulcers usually heal with a medical regimen. Other causes of peptic ulcer disease in children include the Zollinger-Ellison syndrome, cystic fibrosis, short bowel syndrome, and hyperparathyroidism. The Zollinger-Ellison syndrome is rare, occurs mainly in middle-aged adults but has been reported in children, and is characterized by peptic ulceration resistant to therapy, giant gastric rugal folds, and increased serum levels of gastrin. Ulceration due to mucosal injury caused by NSAIDs or other medications is also a diagnostic consideration in older children. Among 360 children with gastritis studied by Dohil et al. (24), 55% had no detectable cause.
Ménétrier Disease
Ménétrier disease is found primarily in adults but is known to occur in children. Though similarities exist, the clinical course and etiology are different. In adults, the cause is unknown and the disease is usually severe and often requires gastrectomy. Childhood cases are often self-limited, and most are caused by CMV infection. Classic Ménétrier disease presents with epigastric or abdominal pain and weight loss. In contrast to adults, children often also present with complications of protein-losing gastropathy, such as ascites, pleural effusions, and periorbital or peripheral edema.
Unlike the adult form, Ménétrier disease in children does not spare the antrum, and hypertrophic gastric folds can be seen throughout the stomach. Histologic features include mucous cell hyperplasia, pronounced elongation and tortuosity of the usually short gastric pits (foveolae), glandular atrophy, and reversal of the usual pit-to-gland ratio. Cysts lined by superficial mucous cells are found deep in the mucosa. Inflammation is more prominent in children than in adults, reflecting the infectious etiology in most children. CMV inclusions are often evident in biopsy material in children. Gastric and urine cultures, serology, polymerase chain reaction and immunohistochemistry, and/or in situ hybridization may be useful ancillary tests. Other associations have included allergy, autoimmunity, and other infections, such as Campylobacter and herpes.
Ménétrier disease is difficult to diagnose in superficial mucosal biopsy specimens. The differential diagnosis includes other causes of large gastric folds: H. pylori gastritis, EG, chronic varioliform gastritis, lymphoma, lymphangiectasia, and other infectious gastritides such as tuberculosis, syphilis, and histoplasmosis.
Foveolar hyperplasia of the antrum in neonates may be caused by prostaglandin therapy administered to maintain patency of the ductus arteriosus in certain forms of congenital heart disease (25). Usually, the clinical setting, antral location, and presence of hypoalbuminemia make it possible to distinguish this group of neonates from those with Ménétrier disease.
Granulomatous Gastritis
Ectors et al. (26) presented the spectrum of causes or associations of granulomatous gastritis in 71 adults. CD constituted 52%; isolated idiopathic granulomatous gastritis, 25%; foreign body granulomas, 10%; and the remainder were related to tumors, sarcoidosis, Whipple disease, vasculitis, or unclassifiable. In our experience, CD is by far the most common cause in children followed remotely by CGD. CGD, an inherited defect of granulocytes, may lead to pyloric outlet obstruction. Endoscopically, in such patients, the antral mucosa is thickened and irregular, and surgical specimens have inflamed mucosa, submucosa, and muscularis. The infiltrate contains mononuclear inflammatory cells, multinucleate histiocytes, and eosinophils. Some have granulomas and foci of necrosis.
Crohn Disease of the Stomach
Involvement of the stomach by CD usually occurs in association with disease in the more common locations—the distal ileum and colon. In a review of 230 children with CD by Lenaerts et al. (27), 30% had lesions of the esophagus, stomach, and duodenum. On occasion, the initial presentation of CD is as a gastroduodenal process. In such cases, the antrum is usually involved, often in continuity with the proximal duodenum. Obstruction of the gastric outlet is a feature shared with EG and some cases of H. pylori gastritis. The histology of gastric CD is similar to that in other sites. Particularly suggestive of gastroduodenal CD is the combination of distinctly focal acute inflammation causing destruction of glandular epithelium plus spotty chronic inflammation similar to the characteristic focal involvement of the distal gastrointestinal tract in CD. This focally enhanced pattern of active gastritis in CD is usually distinct from the more diffuse, superficial and plasma cell predominant pattern of gastritis due to H. pylori infection. In a retrospective study of 238 children with upper gastrointestinal biopsies, focal gastritis was present in 65% of patients with CD and in 20.8% of patients with ulcerative colitis (UC), compared to 2.3% of controls without IBD and one of 39 with H. pylori (28). The presence of granulomas is very helpful in addition to these nonspecific inflammatory features. Pascasio reviewed 438 consecutive biopsies in children with gastritis looking for histologic markers for CD such as granulomas and focal glandulitis (29). Of 58 patients diagnosed as having CD by colonic biopsy and other standard criteria, 34 (77%) were predicted to have CD by gastric biopsy alone. Eosinophils were a significant component in many of the inflammatory foci. In their experience, none of the focal glandulitis biopsies had a history of UC.
Polyps and Tumors of the Stomach
Gastric polyps are rare in children. Juvenile polyps and Peutz-Jeghers polyps may occur in the stomach as part of a generalized polyposis syndrome. Gastric hyperplastic polyps are rare in children but can occur in the setting of H. pylori gastritis. Fundic gland polyps are a more common clinically insignificant consequence of chronic administration of proton pump inhibitors used to treat GERD and dyspepsia. They are usually small and often multiple and are restricted to the oxyntic mucosa of the proximal stomach. Histologically, they can be difficult to distinguish from normal gastric fundic mucosa as the histologic features can be subtle, despite the endoscopic appearance of a polypoid lesion. The diagnostic histologic features include dilatation of the fundic glands and parietal cells with cytoplasmic protrusions extending into the glandular lumina. Cytoplasmic vacuolization of parietal cells is also common. The complete absence of lamina propria inflammation and edema is a striking feature of these polyps (eFigure 14-5). The surrounding flat fundic mucosa often exhibits histologic features similar to but not as pronounced as those evident in the polyps. Fundic gland polyps also develop commonly in patients with familial polyposis coli. Thus, if a fundic gland polyp is identified in a young patient not taking a proton pump inhibitor, colonoscopy to exclude colonic polyposis may be indicated. Dysplasia does occur in fundic gland polyps associated with familial polyposis coli but is exceedingly rare in the sporadic setting (30). For this reason, it is not necessary to remove multiple sporadic fundic gland polyps.
Gastric teratomas are large, bulky multicystic masses that project into the gastric lumen or outward into the peritoneal space. Heterotopic pancreatic tissue should be considered in the differential diagnosis of gastric tumors.
Malignant tumors of the stomach are quite rare in children. MALT lymphomas associated with H. pylori infection and Burkitt lymphomas are the most common types of lymphoma reported (31). Adenocarcinoma of the stomach is distinctly rare but has been reported in otherwise normal children. It is also known to occur in ataxia-telangiectasia and other primary immunodeficiency disorders. Rare examples of inflammatory myofibroblastic tumor and rhabdomyosarcoma have also been reported.
Gastrointestinal Stromal Tumors
Gastrointestinal stromal tumors (GISTs) occur mainly in middle-aged and older patients, with an estimated incidence of about 5000 cases annually in the United States (32). Approximately 85% occur in the stomach, with most of the remainder occurring in the small bowel. Presentation elsewhere in the gastrointestinal tract is unusual. About 1% of GISTs present in children, either as sporadic tumors or in the setting of a syndrome. The vast majority of, but not all, GISTs in children occur in the stomach. Iron deficiency anemia is the most common presenting symptom in sporadic cases, while abdominal pain, a palpable mass, or vomiting occurs rarely (33). Two closely related syndromes feature GIST: the Carney triad and the Carney-Stratakis syndrome. Carney triad is used to describe patients with paragangliomas, pulmonary chondromas, and GIST. About 85% of patients with Carney triad are female, and the GISTs are often multifocal, which is unusual for sporadic tumors. Despite extensive molecular analysis, a specific underlying genetic defect has not been identified in patients with Carney triad (34) Carney-Stratakis syndrome designates a separate group of patients with GISTs and paragangliomas but no pulmonary chondromas. In these patients, autosomal dominant transmission has been demonstrated and germ-line mutations in any of three mitochondrial complex II succinate dehydrogenase (SDH) enzyme subunits (SDHB, SDHC, or SDHD) have been documented (35). GISTs can also develop in individuals with neurofibromatosis type 1, although usually not in childhood.
GISTs are presumed to develop from the interstitial cells of Cajal, which are thought to represent the pacemaker cells throughout the gastrointestinal tract. These cells are normally located within the myenteric plexus and the muscularis propria and have an important role in the regulation of peristalsis. In adults, approximately 90% of GISTs are associated with gain-of-function mutations of the KIT or PDGFR genes. In contrast, these mutations are only present in 15% of pediatric GISTs (32). Thus, the molecular pathogenesis of pediatric GIST is distinct from the adult counterparts, and the underlying mechanisms are currently undefined.
Pediatric GISTs can be of spindle cell or epithelioid morphology, and mixed forms are also common. Among sporadic tumors, epithelioid tumors are more common overall, but spindle cell morphology is more common in boys. The epithelioid tumors are composed of round to polygonal cells, which may have little or abundant cytoplasm. Cytoplasmic vacuolization is common in these tumors and sometimes can be so prominent as to produce a signet ring cell-like appearance (Figure 14-13A, B). The vacuoles do not stain for mucosubstances, glycogen, or fat and appear to represent an artifact of formalin fixation. The spindle cell variant of the tumor resembles smooth muscle tumors (SMT), but the cells are usually not as long and slender. Areas of hyaline fibrosis are common in both spindle cell and epithelioid variants of the tumor (see Chapter 25).
The diagnosis of GIST is confirmed by immunohistologic detection of cytoplasmic reactivity in tumor cells with the c-kit antibody. Even in pediatric tumors where 15% or less of the tumors have mutations in either the c-kit or PDGFRA genes, most of the tumors still express c-kit by immunohistochemistry. In adults, immunohistologic detection utilizing a recently developed antibody-designated DOG1 has been reported as highly sensitive and specific for the diagnosis of GISTs, including those that are nonreactive with the c-kit antibody (36). The antigen detected by the DOG1 antibody is uniformly present in Cajal cells throughout the gastrointestinal tract, but not in mast cells, unlike the c-kit protein (37). In one study, 9 of 11 pediatric GISTs were reactive with the DOG1 antibody (38).
FIGURE 14-13 • Gastric gastrointestinal stromal tumor. A: This example demonstrates epithelioid histology, which is more common in the pediatric age group 100×. B: Cytoplasmic vacuoles are sometimes prominent, as seen here 200×.
In the pediatric population, the differential diagnosis of GIST includes leiomyoma, inflammatory myofibroblastic tumor, desmoid fibromatosis, and monophasic synovial sarcoma. In addition to the greater degree of spindle cell eosinophilia, leiomyoma can be excluded based on immunohistochemical negativity for CD117 and DOG1. Prognostic stratification for GISTs has relied primarily on tumor size and mitotic rate (mitotic figures per 50 HPFs) (39). Management recommendations for pediatric GIST depend on KIT and PDGFRA mutation status as it is presumed that pediatric GIST carrying a KIT or PDGFRA mutation will have a similar evolution and response as adult GIST. In these cases, the mainstay of treatment is imatinib mesylate, an inhibitor that binds to the intracellular portion of KIT and inhibits intracellular signaling. For the majority of children with mutation-negative tumors, the primary treatment is complete surgical resection with the goal of obtaining negative margins, which usually means either a total gastrectomy or a local wedge resection. Adjuvant imatinib is not recommended in mutation-negative GIST as it is not believed to be effective. In the St Jude experience, the incidence of local recurrence was high after primary resection (70%), and greater than 80% after re-resection, with most recurrences manifesting as small peritoneal nodules (35). Because of the usually indolent nature of GISTs in children, surveillance without therapy is recommended for asymptomatic patients with unresectable or metastatic disease.
DISORDERS OF THE SMALL AND LARGE INTESTINE
Congenital Abnormalities
Omphalocele
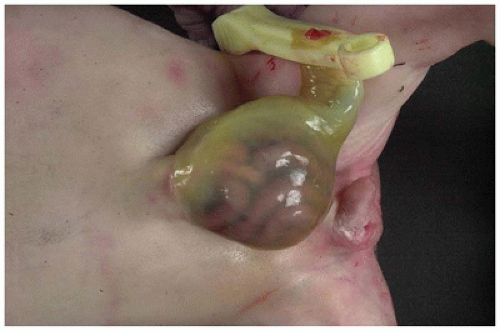
Omphalocele (exomphalos), which has an incidence of about 2 per 10,000 births (40), is a developmental defect of the anterior abdominal wall in which the abdominal musculature, fascia, and skin are absent in the midline at the point of insertion of the umbilical cord. Abdominal organs extrude anteriorly through the defect and are covered by a saclike membrane consisting of amnion externally and parietal peritoneum internally (Figure 14-14). Omphalocele results from failure of the intestine to return to the body cavity after its normal herniation into the umbilical stalk during embryonic life. Omphaloceles vary in size; the defect may be a few centimeters in diameter, or most of the anterior abdominal wall may be lacking. Depending on the size of the defect, small intestine, liver, spleen, and pancreas may be in the sac. The umbilicus usually inserts at the dome of the sac, and umbilical vessels ramify across the membrane. Intrauterine rupture of the sac may occur; exposure of the gut to amniotic fluid results in edema, bowel wall thickening, and matting of intestinal loops. Such cases must be distinguished from gastroschisis. The intestine is nearly always malrotated and shorter than normal.
Other congenital anomalies are found in at least one-third of these infants, including gastrointestinal malformations, congenital heart disease, genitourinary anomalies, imperforate anus, and central nervous system defects. The incidence of omphalocele is increased in infants with trisomy 18, trisomy 13, and trisomy 21. Omphalocele is a key feature of Beckwith-Wiedemann syndrome (gigantism, macroglossia, hemihypertrophy, visceromegaly, and hypoglycemia).
The prognosis in omphalocele is usually determined by the other anomalies and the size of the defect. Emergent operation is not necessary for intact omphaloceles, allowing workup of the neonate for concurrent abnormalities. The sac can be closed primarily if the defect is small or by staged procedures for larger defects using mesh to close the defect (40).
FIGURE 14-14 • Omphalocele. A translucent membrane covers the abdominal organs, which are protruding through an abdominal wall defect in this newborn. Note the insertion of the umbilicus into the center of the omphalocele sac.
Gastroschisis
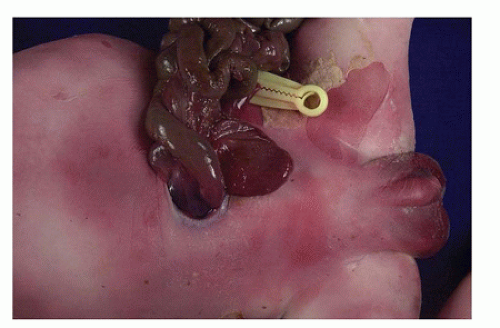
Gastroschisis results from a relatively small, usually rightsided paraumbilical abdominal wall defect, which is distinctly separate from the normally placed umbilicus. Loops of bowel, not covered by a membrane, extrude through the opening (Figure 14-15). Because the extruded intestine has been bathed in amniotic fluid in utero, it appears abnormally thickened and edematous and may be coated with fibrin. The intestine is usually not rotated and is much shorter than normal. Jejunoileal atresia is another recognized association. In contrast to omphalocele, gastroschisis is rarely associated with concurrent major congenital anomalies (41).
The condition has a prevalence of about 1 in 10,000 live births, probably higher if stillbirths and aborted fetuses are included, with a reported increase in the last 30 years (42). Its etiology is unknown; incomplete closure of the lateral folds of the abdomen during development and occlusion or disruption of the right omphalomesenteric artery have been proposed. Maternal factors such as smoking and use of certain medications may play a role (43). Treatment consists of surgical closure of the defect at birth or staged procedures with the use of prosthetic material.
Malrotation
The term malrotation includes a group of congenital positional and associated abnormalities of the intestine and mesentery resulting from nonrotation or abnormal rotation and fixation of the developing embryonic gut. During the most rapid period of growth, the embryonic intestine extends outside the abdominal cavity and initially rotates 90 degrees counterclockwise around the superior mesenteric artery (Figure 14-3). During weeks 10 and 11 of gestation, the intestine returns to the abdomen in sequential stages, with a further 180 degrees counterclockwise rotation until the duodenum comes to rest in its usual position posterior to the superior mesenteric artery. The cecum and right colon, initially located in the right upper quadrant of the abdomen after the return phase, then “descend” into the right lower quadrant anterior to the superior mesenteric artery. At week 11, fixation of the gut to the abdominal wall occurs. A broadbased mesentery extending from the ligament of Treitz to the ileocecal area attaches the intestine to the posterior abdominal wall and stabilizes it. The right and left portions of the colon become fixed retroperitoneally.
FIGURE 14-15 • Gastroschisis. Loops of intestine extrude through an abdominal wall defect located to the right of the normally placed umbilicus. The intestines are not covered by a sac.
Failure of this sequence to take place at all (nonrotation) or failure at any step produces a spectrum of malrotation abnormalities. Any arrest in the process of rotation also tends to interfere with the normal mesenteric fixation of the bowel and results in a narrow mesenteric base and a mobile intestine that is predisposed to volvulus. A person with an incompletely rotated bowel is likely to have abnormal mesenteric fixations and associated extrinsic intestinal obstruction and volvulus. Malrotation often occurs together with other congenital anomalies, including duodenal atresia, omphalocele, gastroschisis, jejunoileal atresia, and Meckel diverticulum.
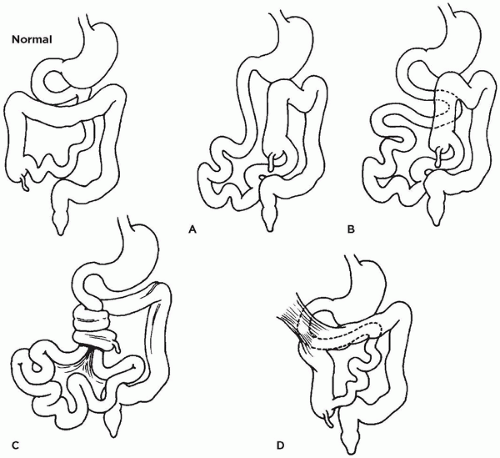
Variations of malrotation are diagrammed in Figure 14-16. In the case of nonrotation (Figure 14-16A), the duodenum is directed inferiorly and lacks the usual sweep to the left. The distal portion of the duodenum and the ascending colon lie together in the midabdomen and are attached to the abdominal wall posteriorly by a very short mesenteric root containing the superior mesenteric artery. The descending colon is not fixed. The narrow mesenteric root and nonfixed descending colon result in midgut volvulus and duodenal obstruction (Figure 14-16C). The rapid progression of volvulus causes the most dreaded and lethal complication of malrotation, which is cessation of mesenteric artery blood flow at the base of the twisted mesentery and infarction of the entire midgut. Midgut volvulus usually presents in the first month of life with intestinal obstruction. Normal rotation of the duodenal loop with nonrotation of the colon is associated with the same potential for midgut volvulus (Figure 14-16B).
FIGURE 14-16 • Normal rotation and variations in position of stomach and intestines due to malrotation. A: Nonrotation of duodenum and colon. B: Nonrotation of colon. C: Midgut volvulus resulting from a narrow mesenteric root and nonfixed descending colon in malrotation. Occlusion of mesenteric blood flow leads to midgut infarction. D: Ladd (peritoneal) fibrous bands (upper left) may extend from the lateral abdominal wall to the right colon, compressing and obstructing the duodenum.
In the variation of normal colonic rotation with nonrotation of the duodenum, abnormal mesenteric bands may intermittently obstruct the duodenum. In another variation, both the duodenum and the colon rotate normally, but the ascending colon does not become fixed. Abnormal peritoneal (Ladd) bands between the hepatic flexure and the right abdominal wall may obstruct the duodenum (Figure 14-16D).
Intestinal Atresia and Stenosis
Intestinal atresia is the complete absence of a segment of the intestine or complete occlusion of the intestinal lumen. Either situation is a common cause of neonatal intestinal obstruction, with a prevalence of 2 in 10,000 live births (44). The rates of atresia in the duodenum and in the more distal jejunum and ileum are approximately equal; colonic atresia is much less frequent. Multiple jejunoileal atresias are found in approximately 10% of cases and can be associated with a number of disorders (44).
Clinical, pathologic, histologic, and experimental observations indicate that most jejunoileal atresias and stenoses are secondary malformations. The disruptions may be caused by intrauterine vascular accidents, with infarction and subsequent resorption or scarring of the affected segment. The fact that bile and squamous epithelial cells are often found distal to the obstruction suggests that the lumen was patent early in gestation. The presence of serosal fibrosis and meconium indicates previous (intrauterine) intestinal perforation and peritonitis. Experimental occlusion of portions of the mesenteric circulation in fetal animals results in identical atretic lesions. Atresias are associated with known vascular insults, such as intrauterine malrotation with volvulus, intussusception, internal hernia, and constricting gastroschisis. Familial patterns in some cases of multiple jejunoileal atresias indicate that not all cases result from vascular accidents. An inherited lethal form of multiple intestinal atresias reported mainly in French Canadians has been associated with mutations in the gene TTC7A located on chromosome 2p21 (45). Examination of the bowel showed sievelike lumina in most of these cases (46).
Approximately 75% of all intestinal stenoses and 40% of all intestinal atresias are duodenal. Intrinsic duodenal atresias and stenoses most often involve the first and second portions, the foregut portion of the duodenum, in close proximity to the entrances of the biliary and pancreatic ducts. This explains the association of duodenal atresia with hepatobiliary and pancreatic duct abnormalities. The embryologic basis of duodenal atresia also differs from that of jejunoileal and colonic atresia. Because most cases of duodenal atresia are of the membranous type, they probably result from a lack of central vacuolization during the solid cord stage of duodenal development. The rate of associated anomalies in infants with duodenal atresia is high. One-fourth of infants with duodenal atresia have Down syndrome, an association not noted with atresia at other sites. Additional congenital anomalies associated with duodenal atresia include cardiac and renal malformations, esophageal atresia, imperforate anus, and vertebral anomalies. Annular pancreas and malrotation are each found in approximately one-fourth of infants with duodenal atresia (47). Jejunoileal atresia is less likely to be associated with other anomalies, although an association has been reported with cystic fibrosis, with an estimated risk factor for cystic fibrosis 210 times higher in Caucasian infants with jejunoileal atresia than those without atresia (48).
The symptoms of intestinal atresia depend on the level of gastrointestinal tract affected. Duodenal and proximal jejunal atresia cause maternal polyhydramnios (secondary to reduced absorption of swallowed amniotic fluid), vomiting, and abdominal distension in the first 24 hours of life; these symptoms are delayed with more distal obstruction. Abdominal radiographs show gaseous distension of the stomach with duodenal atresia and, in lower intestinal atresias, air-fluid levels. Many cases of jejunoileal atresia are now detected by prenatal ultrasonography.
Intestinal atresias have been classified according to their gross appearance (49) (Figure 14-17A). Type I has an intact intestinal wall and mesentery but a septal or membranous luminal obstruction. Because the proximal segment is obstructed, its diameter greatly exceeds that of the distal segment. In type II, two intestinal segments with blind ends are separated by a fibrous cord. Histologic examination of a type II atresia usually shows a recognizable intestinal wall with muscularis layers but fibrous obliteration of the mucosa and submucosa. The frequent presence of luminal granulation tissue, fibrosis, and calcification suggests previous ischemia and healing. In type III, the most common, two blind ends are present without an intervening cord; a wedge-shaped mesenteric defect is also present. Type III may also be associated with a congenitally short small intestine. In the “apple-peel” or “Christmas tree” variety of extensive jejunal atresia, the intestine is very short and the distal ileal segment is coiled around its arterial blood supply (the ileocecal artery) (Figure 14-17B).
Intestinal segments adjacent to regions of atresia or stenosis should be examined for changes suggestive of cystic fibrosis: dilated glands with eosinophilic inspissated secretions, unusually viscid secretions in the lumen, and hyperplasia of goblet cells.
Congenital intestinal stenosis is less common than intestinal atresia; it may be solitary or multiple and may affect a short or long segment. The bowel diameter is greatly reduced, although the lumen is patent throughout. Histologic examination of the intestinal wall often shows evidence of previous ischemia and healing, including mucosal atrophy, submucosal fibrosis, and scarring of smooth muscle. As in intestinal atresia, most cases are presumed to result from an intrauterine ischemic insult, although a history of an untoward event during pregnancy is often lacking.
Duplications of the Gastrointestinal Tract
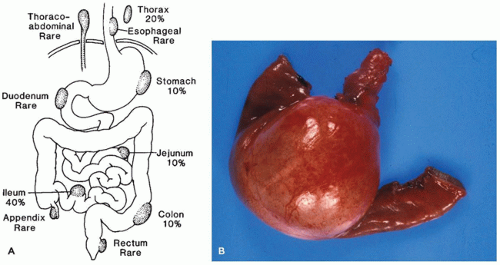
Gastrointestinal (enteric) duplications are tubular or cystic structures that lie alongside the intestinal tube. The duplication and the intestinal tube often share a muscular wall (intramural); less often, the duplication is separated from the intestine proper but in close proximity to it (extramural). Duplications may occur anywhere near the gastrointestinal tract from the neck to rectum; the single most common site is the ileum (Figure 14-18A).
Symptoms vary widely, depending on the location of the duplication. Thoracic enteric duplication cysts are usually extramural and found in the posterior mediastinum; they present with respiratory symptoms in infancy and may communicate across the diaphragm with the intra-abdominal gastrointestinal tract. Abdominal duplications may present with pain, a palpable mass, intestinal obstruction, and, if peptic ulceration occurs in ectopic gastric mucosa, intestinal bleeding.
Multiple duplications are found in 5% of patients. The usual intestinal duplication is a cystic mass located on the mesenteric border. It ranges in size from 2 to 7 cm in diameter, although much larger ones may also be found (Figure 14-18B). The cyst lumen usually does not communicate with the intestinal lumen. Occasionally, tubular duplications paralleling a long segment of intestine are found, usually incidentally. Noncommunicating cysts are filled with mucoid material and histologically mimic normal gastrointestinal tract with enteric mucosa, submucosa, muscularis propria, and a myenteric plexus. Intramural duplications usually do not have a complete muscularis layer but rather share a muscularis layer with the adjacent intestine. The mucosa may resemble adjacent normal gastrointestinal mucosa, but it is often very simplified and difficult to categorize except that columnar epithelium bears a generic resemblance to intestinal surface epithelial cells. Cilia may be present, as in embryonic intestinal epithelium. Gastric mucosa is found in approximately 20% of duplications and may cause peptic ulceration in unlikely sites, such as the ileum and posterior mediastinum. Intestinal duplications in the abdomen must be distinguished from a Meckel diverticulum and other vitelline duct remnants, mesenteric cyst (which lacks intestinal wall morphology), and cystic lymphangioma.
FIGURE 14-17 • A: Classification of intestinal atresia. I: Mucosal (membranous) atresia with intact bowel wall and mesentery; II: blind intestinal ends attached by a fibrous cord; IIIa: blind intestinal ends separated by a V-shaped mesenteric defect without an intervening cord; IIIb: “apple-peel” atresia; and IV: multiple atresia. (From Grosfeld JL. Jejunoileal atresia and stenosis. In: Welch KJ, Randolph SG, Ravich MM, et al., eds. Pediatric Surgery, 4th ed. Chicago, IL: Year Book; 1986:843, with permission.) B: Apple-peel or Christmas tree atresia.
FIGURE 14-18 • A: Locations and incidence of gastrointestinal duplication cysts. B: Cystic duplication of the small intestine.
Meckel Diverticulum and Other Vitelline Duct Anomalies
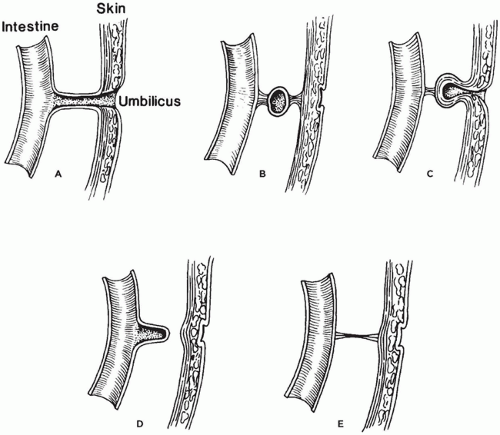
The vitelline (omphalomesenteric) duct usually becomes obliterated by week 10 of embryonic life and subsequently disappears completely. In approximately 2% of the population, however, it remains in various forms (Figure 14-19A to E). These include Meckel diverticulum or, less commonly, a fibrous cord extending from ileum to umbilicus, a cyst, or an umbilical sinus. Many of these remnants are asymptomatic, but others cause symptoms that develop most frequently in the first few years of life.
FIGURE 14-19 • Vitelline (omphalomesenteric) duct anomalies. A: Persistence of the entire vitelline duct from the ileum to umbilicus. B: Vitelline duct cyst. C: Vitelline duct and umbilical sinus. D: Meckel diverticulum. E: Vitelline band.
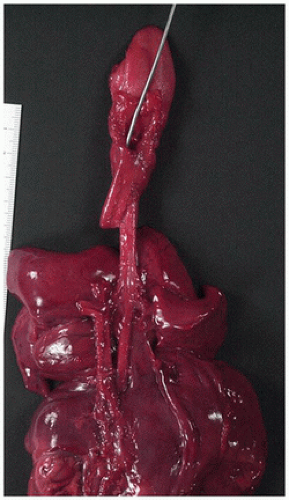
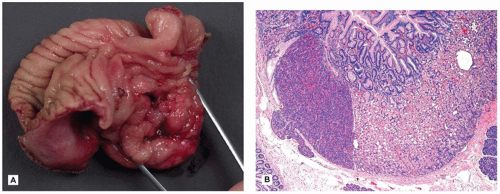
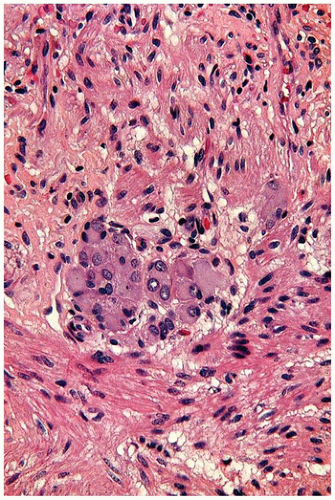
Meckel diverticulum is the most common vitelline duct remnant and also the most common congenital anomaly of the gastrointestinal tract. It results from incomplete obliteration of the vitelline duct at the ileum and appears as a 1- to 5-cm finger-like protrusion of the intestine on the antimesenteric surface of the middle ileum (Figure 14-19D). When found incidentally at autopsy or surgery, most Meckel diverticula are lined by small-intestinal epithelium. Those causing symptoms are likely to contain heterotopic gastric mucosa (Figure 14-20A, B), which secretes acid and leads to peptic ulceration of adjacent intestinal mucosa with subsequent abdominal pain, rectal bleeding, and occasionally intestinal perforation. Approximately 25% of all Meckel diverticula contain foci of gastric mucosa. Occasionally, a Meckel diverticulum may invert into the intestinal lumen and serve as the lead point of an ileal intussusception.
Other vitelline duct remnants are much less common than Meckel diverticulum. A vitelline cyst (Figure 14-19B) results from partial obliteration of the vitelline duct and presents as a mass subjacent to the umbilicus. Microscopically, the cyst wall resembles that of the intestine and is lined by mucus-secreting intestinal epithelium. A vitelline band is a fibrous cord that persists after obliteration of the vitelline duct (Figure 14-19E). These bands extend from umbilicus to ileum, a Meckel diverticulum, or a vitelline cyst and they may serve as a fulcrum for volvulus. Persistence of part of the vitelline duct at the umbilicus causes an umbilical sinus, which presents with mucous discharge from the umbilicus (Figure 14-19C). This must be distinguished from the very rare persistence of the entire vitelline duct (Figure 14-19A). Vitelline cysts and sinuses at the umbilicus are distinguished histologically from urachal remnants at the same site by the presence of intestinal or columnar epithelium. Urachal remnants have a urothelial lining.
FIGURE 14-20 • Meckel diverticulum. A: Gastric mucosa within an opened Meckel diverticulum. B: Ectopic gastric fundic and pancreatic tissue in the mucosa line the diverticulum 100×.
Meconium and Meconium Abnormalities
Meconium is the dark green-to-black mucoid material that fills the neonatal colon and distal small intestine. It consists predominantly of water (75%) admixed with mucous glycoproteins, swallowed vernix caseosa, gastrointestinal secretions, bile, pancreatic enzymes, plasma proteins, minerals, and lipids. More than 90% of healthy term newborns pass a meconium stool averaging 200 mL within the first 24 hours of life, and nearly all have done so by 48 hours. Abnormalities of meconium (e.g., in cystic fibrosis) or of intestinal motility (e.g., in Hirschsprung disease) result in delayed meconium passage.
Meconium Ileus
Meconium ileus is neonatal obstruction of the ileal lumen by abnormally viscid and inspissated meconium containing an abnormally high level of albumin. Most but not all cases occur as the initial manifestation of cystic fibrosis; 10% to 15% of patients with cystic fibrosis are born with meconium ileus. Rarely, infants with congenital pancreatic or pancreatic duct abnormalities have meconium ileus without cystic fibrosis, but these account for fewer than 5% of cases. Thus, the diagnosis of cystic fibrosis should be pursued in every infant with meconium ileus. In the classic case, the distal one-third of the ileum has a nearly normal diameter, but the lumen is filled with dense gray beadlike or solid meconium having the consistency and appearance of putty. The middle one-third of the ileum, proximal to the obstructing meconium, is dilated and filled with dark gelatinous or tarlike meconium. Because the colon in meconium ileus is empty throughout the fetal life, its diameter is smaller than normal.
Microscopically, the distal ileal lumen is filled with hypereosinophilic, focally calcified meconium. Intestinal glands are dilated, often V-shaped, and plugged with hypereosinophilic secretions that are continuous with the luminal meconium. Approximately 50% of the patients with meconium ileus have meconium peritonitis or other complications of meconium ileus, which include intestinal atresia and volvulus.
The overall survival rate of infants with meconium ileus exceeds 80%, although they often have a prolonged hospital course.
Meconium Peritonitis
Intestinal perforation in utero causes meconium to be released into the peritoneal space. Between 33% and 50% of patients with meconium peritonitis have meconium ileus and cystic fibrosis. In the remaining patients, perforation may be the result of intrauterine intestinal obstruction resulting from atresia, vascular insufficiency, malrotation with volvulus, mesenteric hernias, or congenital bands. Meconium peritonitis is usually seen just after birth and is temporally remote from the intrauterine intestinal perforation that caused it. At gross examination, the peritonitis is usually organized, with fibrosis, calcifications, and often dense intestinal adhesions. Occasionally encountered is a meconium pseudocyst, a collection of soft meconium walled off by peritoneal fibrosis.
Microscopically, collections of squames and bile pigment in the peritoneal space, florid fibrosis, and calcifications indicate the presence of meconium. Inflammation is usually chronic, and a well-developed foreign body response to squames and calcifications may be noted. Because the fetal gut is sterile, the degree of inflammation is much less than in the usual case of postnatal peritonitis. If meconium is released into the peritoneal space during intrauterine life, when the inguinal canal to the scrotum is patent, the migration of meconium into the paratesticular area results in a condition called meconium periorchitis (50). Inguinal and even labial meconium masses may also occur, although more rarely, in girls (51).
Meconium Plug
Meconium plug is a syndrome of neonatal colonic obstruction caused by a plug of desiccated meconium, usually in the ascending colon or, in infants with a very low birth weight, the ileum or proximal colon. It is a much less serious condition than meconium ileus, but it may present with a similar clinical picture. The condition may resolve spontaneously, or the plug may pass after a contrast barium enema, and the infant has no further problems. Meconium plug syndrome has been associated with Hirschsprung disease, cystic fibrosis, and maternal tocolysis (52). However, most infants with a meconium plug have none of these conditions.
Gastrointestinal Involvement in Cystic Fibrosis
Cystic fibrosis is a lethal autosomal recessive disease involving multiple organ systems resulting from abnormalities in the gene coding for the cystic fibrosis transmembrane conductance regulator (CFTR), a membrane glycoprotein found in secretory and absorptive surfaces (Chapters 5, 12, and 15). It occurs in approximately 1 in 3500 newborns in the United States (53). Mutations in the gene that encodes CFTR result in faulty electrolyte transport across epithelial surfaces and subsequent dehydration of luminal contents, which in turn leads to obstruction of glands and ducts by thick, viscid secretions. The pancreas, intestines, and lungs are the chief organ systems affected. Gastrointestinal symptoms may be present at birth and almost invariably appear during the first few months of life (Table 14-3). Malabsorption resulting from exocrine pancreatic insufficiency is a prominent manifestation in nearly all children and adults with cystic fibrosis.
Meconium ileus is the first sign of cystic fibrosis in approximately 10% to 15% of patients (54). It usually presents as intestinal obstruction in the first hours or days of life but has also been diagnosed antenatally by obstetric ultrasonography. It should be considered a manifestation of cystic fibrosis until proven otherwise. Up to one-half of infants with meconium ileus have concurrent gastrointestinal manifestations of cystic fibrosis, including meconium peritonitis, smallintestinal atresias and stenoses, duplication, volvulus, microcolon, and mesenteric bands or adhesions.
TABLE 14-3 GASTROINTESTINAL MANIFESTATIONS OF CYSTIC FIBROSIS
Gastroesophageal reflux and Barrett esophagus
Malabsorption and pancreatic insufficiency
Meconium ileus, peritonitis, and meconium plug syndrome
Microcolon
Small-intestinal obstruction and atresia
Intussusception
Volvulus
Rectal prolapse
Brunner gland hyperplasia
Fibrosing colonopathy (due to pancreatic enzyme replacement)
Pneumatosis intestinalis
Mucocele
Gallbladder disease
Distal intestinal obstruction syndrome, formerly called meconium ileus equivalent, is partial or total distal intestinal obstruction by inspissated fecal material occurring in older children and adults with cystic fibrosis. It has nothing to do with meconium. Viscid intestinal contents, a change in dietary habits, dehydration, and temporary disturbances in motility are all thought to play a role. It is more frequent in adults than in children and is associated with more severe genotypes. There is no association with a history of meconium ileus (55). Fibrosing colonopathy is a rare distinctive noninflammatory stricturing process of the colon first described in patients with cystic fibrosis in the 1990s, when it was linked to the ingestion of new preparations of high-dose pancreatic enzyme replacement capsules (56). The condition usually presents with partial or complete intestinal obstruction, and symptoms may mimic those of distal ileal obstruction syndrome (meconium ileus equivalent) or chronic IBD. Fibrosing colonopathy usually affects a long segment of the ascending colon but may involve the entire colon. The lumen is compromised by circumferential submucosal fibrosis along the length of the strictured segment. Fibrosis of the lamina propria, mucosal ulceration, acute and chronic mucosal inflammation, and granulation tissue can also be seen. One series noted increased numbers of eosinophils in the mucosa (57) Current recommendations use lower daily doses of enzyme replacement.
HIRSCHSPRUNG DISEASE
Hirschsprung disease (HSCR, aganglionosis) is characterized by an absence of intramural parasympathetic ganglion cells in the distal gastrointestinal tract resulting in persistent contraction of the affected segment and subsequent colonic obstruction. HSCR is a congenital disorder with an incidence of one per 5000 live births. The condition results from defective craniocaudal migration of vagal neural crest cells (the progenitors of ganglion cells), which populate the entire length of the bowel by the 7th week of gestation. Mutation of one or more HSCR susceptibility genes is the major basis of the disorder (58). More than a dozen genes have been associated with HSCR, the major one being the RET gene, a protooncogene, which codes for a transmembrane receptor, which has also been identified as disease causing in MEN 2A and which maps to chromosome 10q11.2.
More than 90% of patients with Hirschsprung disease are born at full term with a normal birth weight. Presenting symptoms in neonates include delayed passage of meconium (>48 hours), vomiting, abdominal distension, and, in some cases, enterocolitis. In most cases, Hirschsprung disease is an isolated congenital anomaly (70% of patients), but associations have been noted with Down syndrome (10% of patients with Hirschsprung disease have Down syndrome) and other syndromes such as Bardet-Biedl, Mowat-Wilson, multiple endocrine neoplasia (MEN type 2A), Smith-Lemli-Opitz, Waardenburg, and central hypoventilation syndrome (Haddad or Ondine syndrome).
The aganglionic segment in Hirschsprung disease begins at the anal sphincter and extends proximally (Figure 14-21A to E). In 80% of cases, aganglionosis is limited to the rectum and distal sigmoid colon, referred to as short-segment disease. In the remaining patients, the aganglionic segment is longer (long-segment Hirschsprung disease) and extends as far proximally as the splenic flexure or transverse colon in 10% and the cecum in 5% (total colonic aganglionosis or Zuelzer-Wilson disease). In rare cases, aganglionosis extends into the small intestine and may reach as far as the proximal duodenum. Short-segment disease is 5.5 times more common in males, is less likely to recur in siblings, and has features suggestive of multifactorial inheritance. By contrast, long-segment aganglionosis shows a significantly lower male gender bias, higher recurrence rate, and genetic properties suggestive of dominant inheritance with incomplete penetrance (58). In the usual case, barium enema shows a narrow rectum and rectosigmoid colon with a proximal funneling transition to a dilated sigmoid colon. Neonates and infants with long aganglionic segments do not have this diagnostic radiologic picture. Anal manometry is another valuable diagnostic tool used in certain clinical settings. Despite the relative rarity of Hirschsprung disease, it enters into the differential diagnosis of many other conditions because of its varied modes of presentation and the common occurrence of functional constipation in children.
FIGURE 14-21 • Distribution of affected colon in Hirschsprung disease (stippled area). A: Rectosigmoid aganglionosis. B: Ultrashort Hirschsprung disease affected the distal rectum near the anal sphincter. C: Long-segment Hirschsprung disease with involvement of the hepatic flexure. D: Total colonic aganglionosis. E: Aganglionosis involving the entire colon and the distal small bowel (in exceptional cases, the distal duodenum may be involved).
FIGURE 14-22 • Normal submucosal ganglion cells. A: In this 7-month-old child, ganglion cells are easy to identify 400×. B: In this 8-day-old infant, the ganglion cells are immature appearing and therefore more difficult to identify 400×. (Courtesy of Dr. Eduardo Ruchelli, The Children’s Hospital of Philadelphia.)
Although radiographic and manometric studies are routine diagnostic screening procedures, microscopic evaluation of rectal biopsies is the gold standard for the diagnosis of this disorder. The biopsy should be performed at least 1 to 1.5 cm above the dentate line in order to avoid the normal zone of hypoganglionic distal rectum (59). Thus, the first task of the surgical pathologist is to assess the adequacy of the biopsy material. A biopsy that contains squamous or anal transitional epithelium should be reported as inadequate and the absence of ganglion cells in such a specimen is disregarded.
The biopsy must also contain an adequate thickness of submucosa in order to evaluate for loss of ganglion cells. An accepted rule of thumb is that in a well-oriented biopsy, the portion of submucosa sampled should be at least onethird the total cross-sectional area of the biopsy (58). The absence of ganglion cells in an adequate biopsy is diagnostic of HSCR. However, careful examination of a large number of serial sections of each biopsy (75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100) is necessary before a diagnosis of Hirschsprung disease is made. As the average distance between submucosal ganglia may be up to 500 µm, a few sections 3 to 5 µm in thickness may miss the ganglia and result in a false diagnosis of aganglionosis. Further, ganglion cells in neonates can have an immature morphology that can be difficult to recognize in H&E sections, particularly in the submucosa (Meissner plexus), and confusion between endothelial cells and neuronal cells may lead to a false-negative diagnosis (Figure 14-22A, B). One useful feature present in most, but not all, patients with HSCR is the presence of multiple submucosal hypertrophic extrinsic nerve fibers; the presence of nerve fibers with a diameter greater than 40 mm is reported to be characteristic of HSCR (Figure 14-23) (60). Thick submucosal nerve fibers are reportedly less common in short-segment Hirschsprung disease and in total colonic aganglionosis.
Histochemical demonstration of acetylcholinesterasepositive cholinergic nerve fibers within the lamina propria with the presence of thick ropey fibers in the muscularis mucosa provides supportive evidence for the diagnosis of Hirschsprung disease, since they are not present in normal individuals (Figure 14-24). However, this change may not be always evident in patients under the age of 6 months, particularly in short-segment Hirschsprung disease. In addition, this technique can only be performed on frozen sections, requiring the clinician to obtain extra biopsies, and the staining procedure must be followed meticulously using freshly prepared reagents.
Immunohistochemical staining with calretinin has recently become a reliable alternate ancillary method to acetylcholinesterase staining. Calretinin is a calcium-binding protein that is expressed by a subset of submucosal and myenteric ganglion cells, some of which extend neurites into the mucosa. The observation was made that patients with Hirschsprung disease lacked calretinin-immunoreactive submucosal nerve fibers in the aganglionic segment of the colon (61). Absence of calretinin-immunoreactive small nerves or neurites in the lamina propria and muscularis mucosae, with appropriate positive and negative control sections, has now been shown to be the equivalent of AChE histochemistry as a confirmative finding in HSCR (62,63) (Figure 14-25). A clear advantage is that calretinin immunostaining can be performed on formalin-fixed paraffin-embedded sections, obviating the need for additional biopsies or frozen sections. An important caveat that remains to be confirmed in further studies is the presence of calretinin-immunoreactive fibers extending into very short segment of aganglionic colon, complicating the diagnosis of very short segment HSCR (64).
FIGURE 14-23 • Hypertrophic submucosal nerve fibers in a patient with Hirschsprung disease 100×.
FIGURE 14-24 • Acetylcholinesterase stain of a rectal biopsy in a patient with Hirschsprung disease. Note the presence of abnormal ropey nerve fibers in the muscularis mucosae and lamina propria 400×.
FIGURE 14-25 • Calretinin immunostains performed on sections from suction rectal biopsies. A: In a normal infant, calretinin reactive submucosal nerve fibers and neurites in the lamina propria are prominent 200×. B: In an infant with Hirschsprung disease, there is no reactivity in the submucosa or lamina propria in the aganglionic segment 200×.
A number of different histochemical stains (using lactic dehydrogenase, alpha-naphthyl esterase, and NADPH-diaphorase on frozen sections) and different antibodies (S-100, NSE, bcl-2, bone morphogenic protein 1A, and RET) to detect ganglion cells in paraffin sections have been proposed over the years but are not superior to H&E coupled with either acetylcholinesterase or calretinin and thus have not found widespread clinical use.
The definitive treatment of HSCR is surgical, and a number of different operations can be performed. The Swenson and Soave procedures involve removal of the aganglionic segment (in the case of the Soave procedure the mucosa and submucosa of the aganglionic segment are stripped) with creation of a ganglionated neorectum using normally innervated bowel, in either a one-step or two-step procedure with an intervening ostomy. On the other hand, in the Duhamel procedure, ganglionated bowel is brought down and anastomosed directly to the aganglionic segment. In either case, intraoperative seromuscular biopsies for examination of the myenteric (Auerbach) plexus are required mainly to identify normally ganglionated bowel for the anastomosis or ostomy. Seromuscular biopsies should be a minimum of 5 mm in length and require proper orientation and sectioning perpendicular to the serosa to visualize both layers of the muscularis propria to confirm the presence or absence of ganglion cells within the myenteric plexus. Ideally, the specimen should present a shrimplike or C-shaped profile. The myenteric plexus and ganglion cells within seromuscular biopsies are larger and easier to interpret than those in the submucosal plexus, provided the biopsy is adequate and there are no artifacts. In HSCR, nerves are present, usually thicker than normal, and ganglion cells are completely absent. Frozen sections performed above the grossly narrowed segment, to confirm that normal numbers of ganglion cells are present, allow the surgeon to identify the proper level at which to transect the colon. An important point to remember when examining these frozen sections is that colonic segments from patients with HSCR usually contain a transition zone of variable length, with some degree of hypoganglionosis and hypertrophic nerves, immediately proximal to the aganglionic segment. As definitive anastomosis in this zone is associated with continuing postoperative constipation, intraoperative frozen sections must ensure that the segment of bowel used for anastomosis has a normal complement of ganglion cells and normal-caliber nerves (65).
Finally, examination of the resected bowel in HSCR is done to confirm the diagnosis of aganglionosis, determine the length of the aganglionic segment, map the transition zone, and verify the integrity of the innervation of the proximal margin. The latter is best accomplished by examining a complete full-thickness circumference of the margin.
Hirschsprung-associated enterocolitis (HAEC) is a dreaded complication of this disorder and a major cause of morbidity. It can occur before surgical treatment and from 3 weeks to 20 months after the pull-through operation (66) and can develop in aganglionic and ganglionated bowel. Several factors have been associated with an increased risk of developing HAEC: delay in diagnosis, increased length of the aganglionic segment, and trisomy 21. In one series, 45% of patients with trisomy 21 and Hirschsprung disease had enterocolitis (67).
Patients with HAEC present with abdominal distension, vomiting, fever, lethargy, and shock; abdominal radiographs reveal a distended proximal colon and absence of air in the rectosigmoid region, a useful feature, which has been termed the “cutoff” sign (66). Pathologic features can vary from a mild acute cryptitis to a full-blown mucosal ischemic necrosis; some cases may have a pseudomembranous appearance. A grading system for the histopathologic findings has been proposed (68). Neonates may present with a picture suggesting necrotizing enterocolitis (NEC) with pneumatosis intestinalis (69).
The mortality rates vary from 0% to 30%, with lower rates observed in more recent studies (70). It requires aggressive management including antibiotics (71). In cases of repeated enterocolitis, the possibility of persistent obstruction should be investigated, including rectal biopsies to rule out residual aganglionosis.
Chronic Intestinal Pseudo-Obstruction
The term intestinal pseudo-obstruction denotes greatly impaired or absent peristalsis without mechanical obstruction to luminal flow. Pseudo-obstruction is a clinical syndrome, not a pathologic diagnosis, and encompasses a wide spectrum of entities. A classification of these disorders was published recently, the major subclasses of which are neurogenic and myopathic (Table 14-4). The guidelines proposed by this working group also offer diagnostic criteria for these various entities, using both H&E and immunohistochemistry (72).
Visceral neuropathies essentially fall into two main groups: quantitative disorders, where there is an abnormality in the number of enteric neural cells (absent, as in Hirschsprung disease; hypoganglionosis; and hyperganglionosis), and qualitative disorders, where there is a cytomorphologic abnormality. The latter include acquired disorders such as inflammatory and degenerative neuropathies (Chagas disease, lymphocytic and eosinophilic ganglionitis, laxative-induced neurotoxicity) and cytologic abnormalities such as intraneuronal nuclear inclusions, an inherited disorder. The diagnosis of a quantitative abnormality such as hypoganglionosis is less easily accomplished because of a lack of robust quantitative data from normal controls (73). Values for neuronal density appear to vary with age, the region of bowel sampled, the degree of intestinal dilatation, and the methodology. Hypoganglionosis is encountered most frequently in the transition zone proximal to an aganglionic segment. A recent systematic review of isolated hypoganglionosis (not in association with HSCR) identified only 92 published cases, the diagnosis of which remains uncertain given the differences in methodologic workup. Most reported patients presented with some form of slow transit constipation and the outcome has been variable (74). The authors also concluded that diagnosis required full-thickness biopsies. Other techniques have tried to detect abnormalities in the myenteric plexus not otherwise observable on routine H&E sections. For example, silver staining of thick sections of muscularis propria embedded flat to view the myenteric plexus have resulted in the description of a number of rare abnormalities but have not found wide application in clinical diagnosis (75,76). More modern techniques including confocal microscopy and digitalization with three-dimensional viewing of the enteric nervous system offer the possibility of a better understanding of these conditions.
TABLE 14-4 CLINICAL CLASSIFICATION OF PEDIATRIC INTESTINAL PSEUDO-OBSTRUCTION
NEUROPATHIES
Aganglionosis (Hirschsprung disease)
Hypoganglionosis
Congenital
Acquired
Hyperganglionosis
Ganglioneuromatosis
Intestinal neuronal dysplasia
Retarded neuronal maturation
Gliopathies
MYOPATHIES
Muscular malformations
Diffuse abnormal layering of small-intestinal smooth muscle
From Kapur R. Motor disorders. In: Russo P, Ruchelli E, Piccoli D, eds. Pathology of Pediatric Gastrointestinal and Liver Disease. Heidelberg, Germany: Springer Verlag; 2014a:249-317, with permission.
Hyperganglionosis can be encountered mainly in two situations: ganglioneuromas and intestinal neuronal dysplasia (IND). Ganglioneuromas can occur anywhere in the alimentary tract but are most commonly seen in the colon and rectum. The most common form is the isolated polypoid ganglioneuroma, which occurs more frequently in children. These can be sessile or pedunculated, may be grossly indistinguishable from juvenile or hamartomatous polyps, and are usually not associated with disturbances in intestinal motility, neurofibromatosis, or MEN syndromes. On histologic examination, the lamina propria is expanded by a proliferation of ganglion cells and Schwann cells. Multiple ganglioneuromatous polyps (ganglioneuromatous polyposis) are associated with juvenile polyposis and Cowden syndrome (CS). The diffuse form of ganglioneuromatosis is characterized by a proliferation of ganglioneuromatous tissue extensively involving submucosal and myenteric plexuses, which can produce structural thickening along the bowel wall (77). The latter is associated with multiple endocrine neoplasia type 2B (MEN2B), CS, and neurofibromatosis.
IND has been described in association with Hirschsprung disease and as an isolated form. The latter is a controversial entity, with histologic diagnostic criteria that have varied since its initial description 40 years ago, and is neither well described nor widely accepted in the English medical literature. It has been divided into two forms by its proponents: IND-A, the rarest form, and IND-B. Patients with IND-A have been reported to present with constipation since early infancy. Histopathologic descriptions feature hypoplasia of the sympathetic innervation of the myenteric plexus, detected by catecholamine immunofluorescence on frozen sections, in association with hyperplasia of myenteric ganglion cells and increased acetylcholinesterase staining in the lamina propria (78). IND-B, on the other hand, is characterized by the presence of “giant” (>8 ganglion cells) submucosal ganglia (Figure 14-26). There may be associated increased acetylcholinesterase staining of the lamina propria. However, since it is now recognized that giant ganglia are more numerous in premature infants and neonates, this criterion is felt to be diagnostic only in patients older than 4 years of age. Current criteria rely on histoenzymologic stains for dehydrogenases to highlight ganglion cells on 15-µm-thick frozen sections, with “giant” ganglia involving a minimum of 20% of all submucosal ganglia (79). The applicability of these criteria to the evaluation of H&E-stained 4 µm-thick paraffin sections is uncertain. The clinical significance of these findings is equally unclear, and conservative management is considered sufficient, without the need for surgical resection (79). The reproducibility of the histologic diagnosis of IND-B remains poor, and some investigators believe these features may represent a normal variant of postnatal development rather than a pathologic process (80).
FIGURE 14-26 • Giant ganglion in a patient with intestinal neuronal dysplasia 400×.
Visceral myopathies can usually be appreciated by conventional microscopy, although full-thickness specimens of intestinal wall are necessary because the abnormalities are in the muscularis propria. Degeneration, atrophy, and sometimes fibrosis of intestinal smooth muscle are revealed by hematoxylin and eosin stain and enhanced by Masson trichrome stain. The outer, longitudinal layer of the muscularis propria is almost always more affected than the inner, circular layer. Most patients, except those with congenital megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS), present later in childhood or adolescence. MMIHS is a congenital disorder with possible autosomal recessive inheritance diagnosed at birth or by prenatal ultrasonography. Affected infants present with abdominal distension, vomiting, microcolon, and markedly distended bladder and/or ureters. According to a recent review of more than 200 cases, mortality is high, with survivors maintained by parenteral nutrition or bowel transplant (81). Smooth muscle degenerative changes also occur secondarily in the muscular dystrophies, particularly Duchenne muscular dystrophy, in whom esophageal and gastric dysmotility are more common than intestinal pseudo-obstruction. Major pathologic changes include atrophy and fibrosis of the muscularis propria and muscularis mucosa, though few descriptions exist (82). Intestinal dysmotility can also be a key feature of mitochondriopathies, which include mitochondrial neurogastrointestinal encephalomyopathy (MNGIE); mitochondrial myopathy, epilepsy, lactic acidosis, and strokelike episodes (MELAS); and Alpers disease. Pathologic features include patchy atrophy of the muscularis externa and eosinophilic cytoplasmic inclusions in enteric ganglion cells (83). Diffuse abnormal layering of the intestinal smooth muscle is a relatively recently described X-linked disorder with distinctive pathologic features characterized by a trilaminar arrangement of the muscularis propria (84). Other alterations of the visceral smooth muscle include the “brown bowel syndrome,” due to lipofuscin deposition in the muscularis propria resulting from vitamin E deficiency (85) and disorders such as myocyte hypertrophy with honeycomb fibrosis, alpha smooth muscle actin deficiency, and loss of the pacemaker cells of Cajal [reviewed in (58)].
Acquired Diseases
Intussusception
Intussusception, or the invagination of a portion of the intestine into itself, is a relatively common pediatric surgical problem. Infants, particularly those between 5 and 9 months of age, are most commonly affected. More than 90% of cases of childhood intussusception begin at the ileocecal valve, and the intussusceptum may reach as far as the descending colon or rectum. Progressive compression of the blood supply of the invaginated bowel causes edema, hemorrhage, and ischemic necrosis. In the classic case, severe, intermittent, colicky pain begins suddenly in an infant, followed after a few hours by vomiting and the passage of blood and mucus from the rectum. Barium enema is both diagnostic and therapeutic. The obstructing mass of invaginated bowel can be recognized, and if the congestion and edema are not too advanced, the application of hydrostatic pressure by the radiologist reduces the intussusception. Operative reduction is required if barium enema reduction fails, as happens in 20% to 30% of cases (86).
FIGURE 14-27 • Small-bowel intussusception in an infant due to adenovirus infection. A: Histologic section from the lead point of the intussusception demonstrating lymphoid hyperplasia 20×. B: High power demonstrates smudgy intranuclear inclusion within enterocytes 200×. C: Immunostain utilizing an adenovirus antibody confirms the diagnosis 200×.
Gangrene of a portion of the intussusceptum necessitates segmental intestinal resection in approximately 10% of cases. These specimens exhibit edema, congestion, and coagulative and hemorrhagic necrosis indicative of combined ischemia and venous outflow obstruction.
In most cases, the cause is unknown. Large Peyer patches have been proposed as the possible lead point in many cases. Lymphoid hyperplasia due to adenovirus infection has been implicated in a subset of patients (Figure 14-27A to C). Use of the first commercially available oral rotavirus vaccine was associated with an increased risk of intussusception in children, leading to withdrawal of the vaccine from the market (87). Newer-generation vaccines do not appear to have this risk (88).
Approximately 10% of cases of childhood intussusception do not conform to the typical picture. In children past infancy and in atypically located intussusceptions (i.e., those not in the ileocecal valve region), a discrete lead point is usually identified. Meckel diverticulum, Peutz-Jeghers polyps, juvenile polyps, small-intestinal duplications, and Burkitt lymphoma have been implicated.
TABLE 14-5 PEDIATRIC GASTROINTESTINAL INFECTIONS
Organism
Location
Symptom/Syndrome
Histology
Means of Diagnosis
Bacteria
Helicobacter pylori
Stomach, especially antrum
Epigastric abdominal pain in surface
Active chronic nonspecific gastritis; organisms visible mucous coat
Histologic identification of bacilli on biopsy, culture of endoscopic biopsy
Salmonella (S. enteritidis, S. typhi, S. choleraesuis)
Acid-fast bacilli in macrophages throughout lamina propria
Identification of acidfast bacilli in macrophages in lamina propria of intestinal biopsy
Fungi
Candida
All GI tract
Depends on location
Pseudohyphae and yeast forms with acute inflammation
Fungal stain on biopsy
Protozoa
Giardia lamblia
Proximal SI
Diarrhea, malabsorption, failure to thrive
Proximal SI changes range from minimal change to chronic inflammation and villous atrophy. Organisms on H&E
Stool examination for cysts; mucus smears of intestinal biopsy; identification visible of trophozoites on proximal SI biopsy
Cryptosporidium
Small intestine in normal hosts; stomach, SI, and colon in AIDS patients
Watery diarrhea
Minimal change or mild, nonspecific enteritis; organisms visible on H&E
Stool examination for cysts; identification by histologic examination of proximal SI biopsy
Entamoeba histolytica
Colon
Hematochezia, diarrhea, bloody, mucoid diarrhea
Diffuse acute colitis, microulcerations, deep ulcers undermining mucosa, organism visible on H&E
Stool examination for trophozoites and cysts; identification of trophozoites on histologic examination of colonic biopsy; serology; identification of organism on biopsy