Oral, Maxillofacial, Head and Neck Pathology in Pediatrics
M. John Hicks, M.D., D.D.S., M.S., Ph.D.
INTRODUCTION
The oral and maxillofacial and head and neck region is composed of vastly different tissue types and experience many of the conditions afflicting specific tissues. However, this region also has unique lesions that are not seen at other body sites or with other organ systems. This chapter will review those entities that are unique to the oral and maxillofacial and head and neck region. These include infectious, reactive, congenital, autoimmune, and benign and malignant neoplastic processes that occur in the pediatric age group.
NASOPHARYNGEAL ANGIOFIBROMA (JUVENILE NASOPHARYNGEAL ANGIOFIBROMA)
Nasopharyngeal angiofibroma (NPA) is a rare tumor representing less than 1% of all head and neck tumors. It occurs exclusively in males in the second decade of life and is uncommon after 25 years of age (1,2,3,4,5,6,7,8). If NPA is diagnosed in a phenotypic female, chromosomal studies are indicated to determine if gonadal dysgenesis is present. The most common symptoms are nasal congestion and epistaxis. Nasal discharge, sinusitis, headache, midface and nasal bridge swelling, proptosis, visual distortion, anosmia, and cranial nerve deficits, including hearing loss, may also occur. Symptoms may be long-standing (1 year or more), but without pain. The most common site of involvement is the posterolateral nasal roof near the sphenopalatine foramen. The tumor may extend anteriorly into the nasal cavity, posteriorly into the oropharynx and nasopharynx, through the sphenopalatine foramen into the pterygomaxillary and infratemporal fossa, and into the middle cranial fossa.
The overwhelming predilection for male gender with NPA is related to the presence of androgen receptors and lack of estrogen receptors, with dependency on testosterone for tumor growth and inhibition by estrogen (1,2,3,4,5,6,7,8). This tumor is also associated with familial adenomatous polyposis syndrome with a 25-fold increased risk. Beta-catenin-activating mutations in the stromal cells are noted within tumors associated with FAP; however, in certain sporadic cases, these mutations may also be present. This emphasizes the need for constitutional testing for beta-catenin mutation if this mutation is detected in the tumor. NPA stromal cells may also express neural growth factor and CD117 (see Chapter 25).
Diagnostic imaging typically shows a soft tissue tumor (Figure 22-1) that is markedly hypervascular with prominent arteries. The feeding vessels for this tumor are most often from the maxillary artery. The vascularity is best appreciated on angiography. Because of the prominent vascularity, embolization of the tumor is often performed prior to biopsy or surgical intervention to avoid excessive blood loss and also to avoid consumptive coagulopathy. The tumor typically shows impingement on and bowing of the posterior wall of the maxillary sinus and posterior displacement of the pterygoid plates (Holman-Miller sign).
NPAs may have several different clinical appearances, such as sessile, lobulated, polypoid, or pedunculated (1,2,3,4,5,6,7,8). Typically, the specimen submitted for gross examination is markedly fragmented and the clinical appearance cannot be appreciated. Microscopic examination shows a tumor with no apparent capsule that is comprised of stroma that is typically of low cellularity with collagen in the background with variably sized thin vessels (Figure 22-1). The vessels have lumens that vary from gaping to stellate to staghorn to poorly visible due to compression by the stromal component. The vessels are lined by typical endothelial cells and have an inconspicuous to incomplete smooth muscle layer. Tumors of long duration tend to have increased collagen content and fewer vessels.
Treatment of NPAs is usually surgical, since spontaneous regression is rare (1,2,3,4,5,6,7,8). Use of testosterone receptor inhibitors, estrogen therapy, and irradiation have been utilized as nonsurgical therapies, especially in nonresectable tumors. Recurrence rates vary from 5% to 25% being higher for those involving the skull base and with extension into the cranium.
There is some degree of mortality associated with NPAs (2% to 10%). Irradiation of these tumors has been rarely associated with sarcomatous transformation and usually with massive radiation dosages.
There is some degree of mortality associated with NPAs (2% to 10%). Irradiation of these tumors has been rarely associated with sarcomatous transformation and usually with massive radiation dosages.
 FIGURE 22-1 • Nasopharyngeal angiofibroma. A: Soft tissue mass (arrow) obstructing nasal cavity and extending into nasopharynx on CT scan. B: Low-cellularity tumor with abundant stroma, prominent gaping vessels lacking muscular walls, and bland slightly spindled stromal cells. |
NASOPHARYNGEAL CARCINOMA, TYPE II NONKERATINIZING, UNDIFFERENTIATED (WHO GRADE TYPE II UNDIFFERENTIATED)
The WHO classification system divides nasopharyngeal carcinoma (NPC) into keratinizing type I, nonkeratinizing type II differentiated, and nonkeratinizing type II undifferentiated (9,10,11,12,13,14,15,16,17,18). This tumor arises from the nasopharyngeal mucosa and there is evidence of squamous differentiation. The most common population affected by this tumor is Asian (southern China and Taiwan) and with decreased risk with immigration to low prevalence areas. However, Chinese descent still carries an increased risk compared with non-Chinese descent. Other ethnic groups commonly afflicted with this tumor include Southeast Asians, central and northern Africans, and Arctic natives. In China, NPC accounts for almost 20% of all cancers. Nonkeratinizing type II undifferentiated NPC represents greater than 60% of cases, while type 1 keratinizing NPC (25%) and type II nonkeratinizing differentiated NPC (<15%) are less frequent. HLA types A2, B17, Bw46, and BW58 may be predisposing factors in NPC tumorigenesis. Pediatric NPCs account for less than 20% of all type II undifferentiated subtypes, with only 2% of NPCs occurring in children in China and up to 20% of NPCs occurring in children in central and northern Africa. In contrast, NPC in the United States accounts for less than 0.5% of all cancers. With the pediatric population, only type II nonkeratinizing undifferentiated tumors occur and have a strong association with Epstein-Barr virus. There is a trend to rename this tumor as Epstein-Barr virus-associated carcinoma.
The tumor is more common in males and typically involves the lateral wall of the nasal cavity (Rosenmüller fossa) and the superior-posterior wall (9,10,11,12,13,14,15,16,17,18). Often, the tumor presents as a neck mass involving the posterior cervical or superior jugular lymph node chain. The nasopharynx mass may be discovered after needle core biopsy (Figure 22-2) or excisional biopsy of a neck lymph node, because presentation is usually at an advanced clinical stage, with some having cranial nerve involvement (up to 25%). The nasopharyngeal mass may cause nasal obstruction, epistaxis, otitis media, hearing loss, and nasal discharge.
Type II undifferentiated NPCs are characterized by large islands of tumor cells surrounded by a lymphocytic infiltrate in the background (9,10,11,12,13,14,15,16,17,18). The nonkeratinizing
undifferentiated tumor cells have minimal cytoplasm with indistinct borders, imparting a syncytial appearance (Figure 22-2). The tumor cells have large oval vesicular nuclei with prominent nucleoli. The tumor cells immunoreact with EBER-1, while the adjacent lymphocytes are negative. The tumor is also negative for p16. The tumor is also positive for pancytokeratin and high molecular weight keratins. EBV of a clonal nature can be identified, indicative of a strong oncogenic association between EBV and nonkeratinizing NPC, especially the undifferentiated subtype. Serology is positive for EBV viral capsid antigen (VCA) and EBV early antigen in 90% of cases. In addition, circulating EBV DNA in both plasma and serum is noted in over 95% of those affected by nonkeratinizing NPC. Oncologic management involves radiation therapy and adjuvant chemotherapy. Clinical stage at diagnosis is the primary factor in survival. Stage I disease has a 98% 5-year survival rate, while stage IV disease has only a 73% 5-year survival rate.
undifferentiated tumor cells have minimal cytoplasm with indistinct borders, imparting a syncytial appearance (Figure 22-2). The tumor cells have large oval vesicular nuclei with prominent nucleoli. The tumor cells immunoreact with EBER-1, while the adjacent lymphocytes are negative. The tumor is also negative for p16. The tumor is also positive for pancytokeratin and high molecular weight keratins. EBV of a clonal nature can be identified, indicative of a strong oncogenic association between EBV and nonkeratinizing NPC, especially the undifferentiated subtype. Serology is positive for EBV viral capsid antigen (VCA) and EBV early antigen in 90% of cases. In addition, circulating EBV DNA in both plasma and serum is noted in over 95% of those affected by nonkeratinizing NPC. Oncologic management involves radiation therapy and adjuvant chemotherapy. Clinical stage at diagnosis is the primary factor in survival. Stage I disease has a 98% 5-year survival rate, while stage IV disease has only a 73% 5-year survival rate.
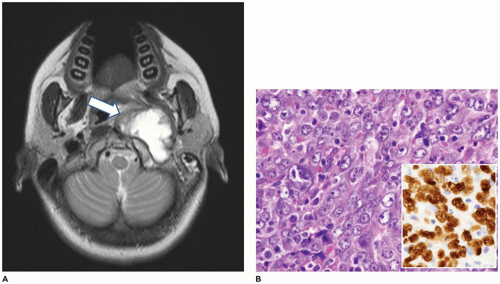 FIGURE 22-2 • Nasopharyngeal carcinoma, type II nonkeratinizing, undifferentiated. A: Soft tissue mass (arrow) involving and expanding into posterolateral aspect of nasopharynx on CT scan. B: Malignant tumor cells with vesicular nuclei, prominent nucleoli, and moderate cytoplasm. Inset: Tumor cells with diffuse nuclear reactivity for EBV (in situ hybridization for EBER-1). |
NASAL CHONDROMESENCHYMAL HAMARTOMA
Nasal chondromesenchymal hamartoma (NCMH) is a rare neoplasm involving the sinonasal region and is comprised of chondroid and stromal components (19,20,21). This lesion is most commonly identified in the neonatal period and usually by 3 months of age. It is more commonly seen in males. The presenting symptom is respiratory difficulty associated with facial swelling and a sinonasal mass, with involvement of the septum and middle turbinate being most common (Figure 22-3). The tumor may erode the cribriform plate and extend into the cranial cavity, and this may mimic a meningoencephalocele. The mean size is 3.6 cm with some tumors of up to 8 cm.
The tumor is characterized by nodules and irregular islands of hyaline and fibrocartilage that are surrounded by bland stromal spindle cells (19,20,21). The stroma may have a myxoid to loose to dense to fibrous character. There is an abrupt transition from the stromal spindle cells to the cartilaginous nodules. These cartilaginous nodules may vary from chondromyxomatous (vaguely resembling chondromyxoid fibroma) to well-differentiated cartilage. There may be several patterns present, such as myxoid spindle cell stroma, fibroosseus proliferation with cellular stroma and immature woven bone mimicking fibrous dysplasia, trabecular and ossicle-like bone formation, and cyst-like blood-filled spaces mimicking aneurysmal bone cyst. Immunohistochemical studies show S100 positivity with the cartilaginous component and smooth muscle actin (SMA) and vimentin reactivity with the stromal spindle cell component.
More recently, there has been an association with DICER-1 mutations and DICER-1 tumor predisposition syndromes, such as familial pleuropulmonary blastoma predisposition
syndrome (19,20,21). The majority of NCMH are nonsyndromic in nature; however, constitutional DICER-1 mutation analysis should be performed in the affected individual.
syndrome (19,20,21). The majority of NCMH are nonsyndromic in nature; however, constitutional DICER-1 mutation analysis should be performed in the affected individual.
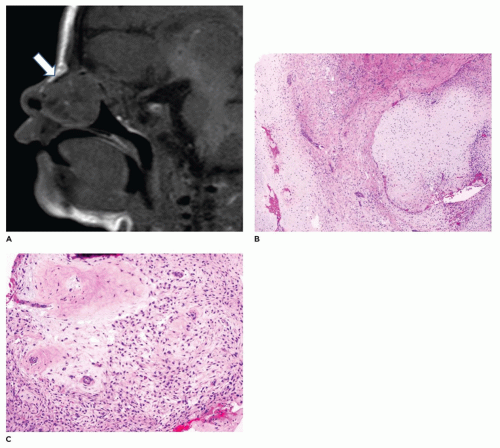 FIGURE 22-3 • Nasal chondromesenchymal hamartoma. A: Well-defined soft tissue mass (arrow) occupying the nasal cavity on CT scan. B, C: Moderately cellular myxofibrous stroma with abrupt transition into cartilaginous nodules. |
NASAL-ASSOCIATED LESIONS
Sinonasal Inflammatory Polyps
With sinonasal inflammatory polyps in general, these polyps most often arise from the lateral nasal wall or from the ethmoid sinus (22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39). These polyps can be single, unilateral, bilateral, or multiple. The typical symptoms are rhinorrhea, nasal cavity obstruction, and headache. Allergies, cystic fibrosis, infections, diabetes, and aspirin sensitivity are often associated with sinonasal polyps. Most occur in individuals older than 20 years of age and infrequently in young children (<5 years of age). However, nasal polyps may be the first sign of cystic fibrosis in young children and typically present in the first or second decade of life in about 20% of affected individuals (Figure 22-4). Polyps in cystic fibrosis contain fewer eosinophils and lack basement membrane thickening. The inspissated mucin in cystic fibrosis is acidic in nature and may be highlighted with PAS-Alcian blue with the mucin staining blue to purple compared with typical mucin that stains pink to magenta.
Sinonasal polyps have a myxoid to gelatinous stroma with a mixed acute and chronic inflammatory infiltrate comprised predominantly of lymphocytes and eosinophils. There is a markedly thickened basement membrane beneath the respiratory epithelium, which may demonstrate squamous metaplasia. Charcot-Leyden crystals may be seen with
abundant eosinophils. Treatment is surgical excision with identification of the underlying cause in order to avoid recurrence.
abundant eosinophils. Treatment is surgical excision with identification of the underlying cause in order to avoid recurrence.
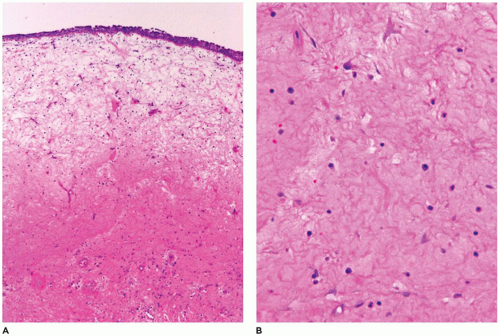 FIGURE 22-4 • Sinonasal polyp in cystic fibrosis. A and B: Intact respiratory mucosa overlying submucosa with abundant deeply eosinophilic mucinous stroma. |
Antrochoanal polyps (AP) arise from the maxillary antrum and are considered sinonasal polyps, which are inflammatory, noninfectious polyps (22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39). APs represent about 5% of sinonasal polyps. Males are more often affected than females and most occur at a young age. Most are unilateral, single polyps (90%) and result in nasal cavity obstruction; and if the AP extends posteriorly, the nasopharynx may be obstructed. These polyps are often seen in conjunction with maxillary sinusitis as well as allergic conditions (up to 40%). Antrochoanal polyps typically show prominent vascular spaces in a fibromyxoid to fibrous background without an inflammatory infiltrate (Figure 22-5). The treatment of APs is surgical with excision of the stalk in order to avoid recurrence.
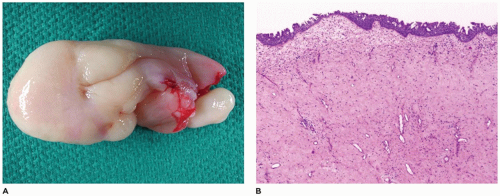 FIGURE 22-5 • Antrochoanal polyp. A: Polyp with intact mucosa and irregular surface contour. B: Intact respiratory mucosa with underlying submucosa comprised of fibromyxoid tissue with prominent vessels. |
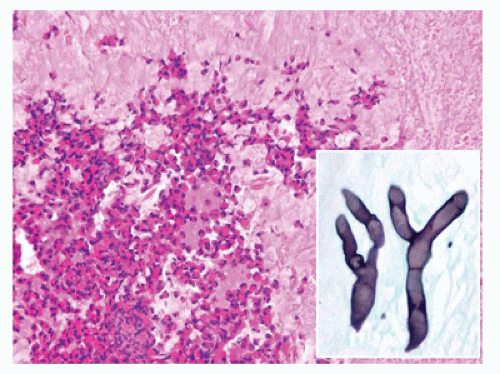 FIGURE 22-6 • Allergic sinusitis. Abundant eosinophilic mucin with aggregates of numerous eosinophils. Inset: Hyphae with 45-degree branching and septations (MSNsilver histochemical stain). |
Sinonasal Mycotic Sinusitis
Sinonasal mycotic sinusitis may be subtyped as acute fulminant, angioinvasive sinusitis, chronic noninvasive indolent sinusitis, mycetoma, or allergic sinusitis. The most common fungal sinusitis is allergic in nature (22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39). Allergic sinusitis occurs in immunocompetent individuals and is a noninvasive fungal sinusitis. The affected individual typically has elevated IgE, peripheral eosinophilia, and a history of a long-standing allergic condition (atopy). This condition is caused by dichotomous fungal hyphae with most organisms in the Dematiaceae family (Bipolaris, Curvularia, Exserohilum, Alternaria, Cladosporium). Microscopic examination shows abundant pale eosinophilic to basophilic mucin with a laminated appearance with numerous eosinophils and other chronic inflammatory cells (Figure 22-6). Fungal organism with 45 degree branching and rare yeast forms may be seen on silver or PAS staining. Mycotic cultures are necessary to determine the precise organisms responsible for the sinusitis. With noninvasive sinusitis, curettage of involved sinus mucosa with systemic antimycotic agents is necessary.
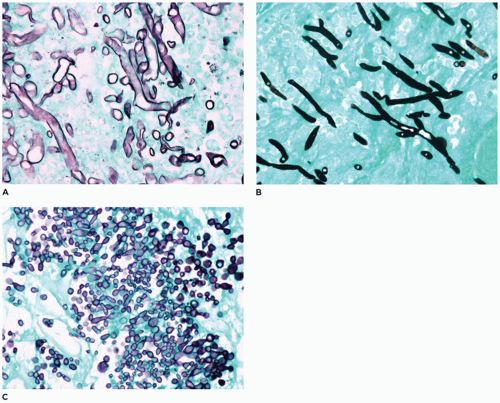 FIGURE 22-7 • Sinonasal mycotic infection with organisms often associated with angioinvasion. A: Zygomycetes species. B: Aspergillus. C: Candida (MSN-silver histochemical stain). |
Acute fulminant angioinvasive mycotic sinusitis is associated with immunocompromised or immunosuppressed hosts, often affecting individuals with hematologic malignancies (22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39). These mycotic infections are characterized by mucosal ulceration, tissue necrosis, acute inflammatory infiltrates, and angioinvasion by organisms (Figure 22-7). Zygomycetes (Mucor), Aspergillus, Candida, Cryptococcus, Curvularia, and
Alternaria may be the invasive mycotic agents. Treatment involves aggressive sinonasal debridement and intravenous antimycotic agents. Survival for invasive mycotic infections is variable from 75% for individuals with no underlying disease to 20% for individuals with leukemia or renal disease. With diabetic patients, the survival rate is almost 80% when appropriate antimycotic agents are utilized.
Alternaria may be the invasive mycotic agents. Treatment involves aggressive sinonasal debridement and intravenous antimycotic agents. Survival for invasive mycotic infections is variable from 75% for individuals with no underlying disease to 20% for individuals with leukemia or renal disease. With diabetic patients, the survival rate is almost 80% when appropriate antimycotic agents are utilized.
NASAL GLIAL HETEROTOPIA (NASAL GLIOMA)
Glial heterotopia is a nonneoplastic, congenital displacement of neuroglial tissue at an extracranial site (40,41,42,43,44). It is often considered to be an encephalocele variant without communication with the central nervous system and not a neoplasm. This entity presents at birth or within the first few weeks of life and is most common in nasal subcutaneous tissues (60%), within the nasal cavity (30%) and less often in the sinuses, palate, middle ear, tonsils, or pharynx. Diagnostic imaging is mandatory to exclude communication with the central nervous system and exclude encephalocele with cranial bony defect. These lesions may present as red to blue area on the nasal bridge. The nasal cavity lesions present with symptoms of obstruction, epistaxis, respiratory distress, deviation of the septum, rhinorrhea from CSF fluid, or even meningitis. The tissue is comprised of an admixture of neuroglial elements, including astrocytes, gemistocytic astrocytes, glial fibers, and fibrovascular tissue (Figure 22-8). Neuronal elements tend to be rare to absent, but some cases have shown abundant neuronal elements. Fibrosis may be more pronounced with long-standing lesions. Immunohistochemical staining is positive for S100 protein and glial fibrillary acidic protein. Treatment is simple surgical resection. If the lesion is determined to be an encephalocele, craniotomy with repair of the bony defect is also necessary. Recurrence is rare if the lesion is completely excised.
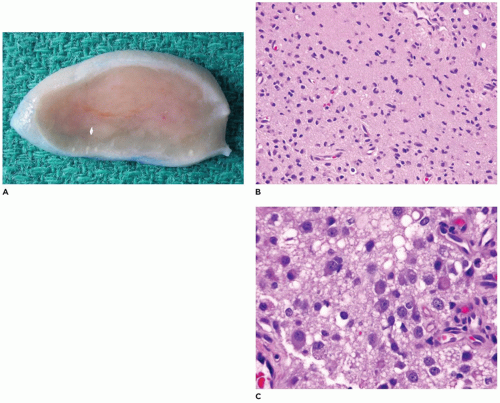 FIGURE 22-8 • Nasal glial heterotopia. A: Sectioning of lesion with intact overlying skin and underlying myxoid tissue. B: Neuroglial tissue with prominent neuropil. C: Neurons within a vacuolated neuroglial background. |
SQUAMOUS PAPILLOMATOSIS, LARYNGOTRACHEAL TYPE (RESPIRATORY PAPILLOMATOSIS)
Laryngeal squamous papillomatosis is associated with multiple lesions involving most commonly the larynx (95%) and less commonly the tracheobronchial tree (5%) (45,46,47,48,49,50).
The association with human papillomavirus (HPV) types 6 and 11 is well known. HPV type 11 is more often associated with more extensive involvement. These lesions occur in two separate forms: juvenile form before 5 years of age and adult form between 20 and 40 years of age. The juvenile form is associated with HPV transmission from the genital lesions of the mother during vaginal delivery. Of note, HPV infection is not completely obviated by cesarean section delivery. There is no gender bias with the juvenile form; however, the adult form affects males more often. The presenting signs and symptoms include inspiratory stridor, hoarseness, and less often obstructive symptoms. With the juvenile form, the lesions tend to recur and grow rapidly even after surgery. The lesions may undergo spontaneous regression during puberty.
The association with human papillomavirus (HPV) types 6 and 11 is well known. HPV type 11 is more often associated with more extensive involvement. These lesions occur in two separate forms: juvenile form before 5 years of age and adult form between 20 and 40 years of age. The juvenile form is associated with HPV transmission from the genital lesions of the mother during vaginal delivery. Of note, HPV infection is not completely obviated by cesarean section delivery. There is no gender bias with the juvenile form; however, the adult form affects males more often. The presenting signs and symptoms include inspiratory stridor, hoarseness, and less often obstructive symptoms. With the juvenile form, the lesions tend to recur and grow rapidly even after surgery. The lesions may undergo spontaneous regression during puberty.
The lesions occur as papillomatous exophytic, pedunculated, or sessile 1- to 5-mm masses along the respiratory mucosa (45,46,47,48,49,50). There is a tendency to bleed upon manipulation and trauma. Microscopic examination shows papillary lesions lined by stratified squamous epithelium with basal cell hyperplasia and with an underlying fibrovascular core (Figure 22-9). Koilocytic changes secondary to HPV infection are noted in the more superficial layers of the epithelium. The surface of the epithelium may demonstrate minimal parakeratinization. Only rarely is dysplasia of the squamous epithelial cells noted (50).
 FIGURE 22-9 • Squamous papillomatosis (A, B) and squamous cell carcinoma (C, D). A: Squamous papilloma with marked squamous cell proliferation with fibrovascular core. B: Koilocytic change with squamous epithelial cells, indicative of HPV infection. C: Squamous papillomatous lesion with superficial squamous cell carcinoma invasion (arrows) into underlying tissue. D: Invasive squamous cell carcinoma infiltrating peribronchial (arrows) and lung tissue. |
Extralaryngeal spread does occur in about one-third of children, and this is most common to the oral cavity and tracheobronchial tree (45,46,47,48,49,50). Endobronchial and lung involvement is noted in about 5% of individuals. Rarely invasive squamous cell carcinoma involving the lung develops in the pediatric age group (Figure 22-9). A poor prognostic factor is development of squamous papillomatosis in the
neonatal period, often necessitating tracheotomy, and may result in demise of the child. There are no other prognostic clinical or pathologic factors that predict the clinical course. Of note, radiation therapy is associated with an increased risk for malignant transformation (see Chapter 12).
neonatal period, often necessitating tracheotomy, and may result in demise of the child. There are no other prognostic clinical or pathologic factors that predict the clinical course. Of note, radiation therapy is associated with an increased risk for malignant transformation (see Chapter 12).
CONGENITAL EPULIS OF THE NEWBORN (GINGIVAL GRANULAR CELL TUMOR OF INFANCY, NEUMANN TUMOR)
Congenital epulis of the newborn is a rare mucosal tumor that is predominantly located on the anterior alveolar ridge of newborns with the maxilla more commonly involved (75%) (51,52,53). The vast majority occur in female neonates (90%) with multiple lesions occurring in 10% of affected neonates. This tumor is typically pedunculated and varies in maximum dimension (0.5 to 9 cm, most 0.5 to 2 cm). The lesion is painless and does not increase in size after being discovered. Some small lesions tend to regress spontaneously over time. Microscopic examination shows intact squamous mucosa with the underlying submucosa filled with markedly enlarged polygonal-shaped tumor cells (Figure 22-10). These tumor cells have abundant granular eosinophilic cytoplasm with bland nuclei. The tumor cells are closely apposed with minimal fibrous stroma and occasional fine vascular channels. The granular cells are negative for S100 protein, CD57, cytokeratin, actin, desmin, estrogen and progesterone receptors, and neuron-specific enolase. Other neural markers are also negative. The granular cells react with vimentin and CD68. Ultrastructural examination shows lysosome, lipid, and variably sized dense granules. The overlying squamous mucosa is intact but lacks pseudoepitheliomatous hyperplasia, which is seen in adult granular cell tumors. Odontogenic epithelial rests interspersed among the granular cells may be seen in up to 50% of these lesions. The tumor cells are believed to be derived from primitive mesenchyme from the neural crest; however, there is no convincing evidence for this belief. Treatment is surgical excision in order to provide a definitive diagnosis and eliminate other entities from concern. Recurrence is extremely rare after even incomplete excision. This is no doubt due to the tendency for some lesions to undergo spontaneous regression (see Chapter 25).
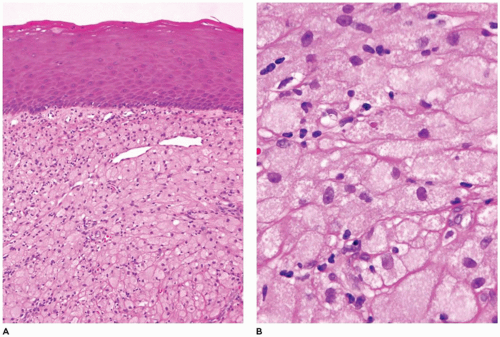 FIGURE 22-10 • Congenital epulis of newborn. A and B: Gingiva with intact stratified squamous mucosa overlies a proliferation of markedly enlarged polygonal cells with lightly eosinophilic granular cytoplasm. |
GIANT CELL LESION OF JAW (CENTRAL AND PERIPHERAL GIANT CELL GRANULOMA)
Giant cell lesion of the jaw is histopathologically and immunohistochemically indistinguishable from its long bone counterpart (54,55,56,57,58). This tumor may occur within the maxilla, mandible, or oral soft tissues without bony involvement.
Although not proven by cytogenetic or molecular techniques, this tumor is most likely a neoplastic one, similar to giant cell tumor of bone. Clonality with t(1;17;18) and other random numerical chromosomal changes have been reported in isolated cases. The age range for this tumor is wide, but about two-thirds occur before age 30 years with a peak in the second decade of life during mid to late adolescence. There is a female predilection. The anterior mandible is most commonly involved and often crosses the midline of the mandibular symphysis. Most tumors are asymptomatic and are discovered during routine dental radiologic examination or because of painless bony expansion. The minority of tumors are rapidly growing aggressive lesions with bone perforation, tooth root resorption and displacement, and paresthesia. Based upon these clinical features, the tumors have been divided into nonaggressive and aggressive lesions.
Although not proven by cytogenetic or molecular techniques, this tumor is most likely a neoplastic one, similar to giant cell tumor of bone. Clonality with t(1;17;18) and other random numerical chromosomal changes have been reported in isolated cases. The age range for this tumor is wide, but about two-thirds occur before age 30 years with a peak in the second decade of life during mid to late adolescence. There is a female predilection. The anterior mandible is most commonly involved and often crosses the midline of the mandibular symphysis. Most tumors are asymptomatic and are discovered during routine dental radiologic examination or because of painless bony expansion. The minority of tumors are rapidly growing aggressive lesions with bone perforation, tooth root resorption and displacement, and paresthesia. Based upon these clinical features, the tumors have been divided into nonaggressive and aggressive lesions.
Biopsy of the lesion is performed prior to surgical intervention (54,55,56,57,58). The tumor is comprised of giant cells of variable sizes and shapes with varying numbers of nuclei per cells (up to 20 per cell), resembling osteoclast-like giant cells (Figure 22-11). In the background are mononuclear stromal cells, which vary from round to oval or spindle shaped. The stromal cell nuclei are similar in appearance to the giant cell nuclei. The mononuclear stromal cells may be seen merging and fusing with the giant cells. Immunohistochemical staining with CD68 highlights the cytoplasm of both the stromal cells and giant cells, suggesting a common histiocytic derivation. There is also a fine vascular pattern within the stroma. Cytogenetic and molecular genetic studies have not been analyzed thoroughly with this tumor. However, the SH3BP2 gene mutation, which leads to RANKL-induced activation of calcineurin and NFAT proteins responsible for the giant cell lesion phenotype in cherubism, has not been identified in giant cell lesions of the jaw (59). This indicates that giant cell lesions of the jaw are a distinct entity from the lesions associated with cherubism and have a different pathogenesis. Ramon, Noonan, Schimmelpenning, and neurofibromatosis type 1 syndromes are inherited conditions that have been associated with giant cell lesions of the jaw (54,55,56,57,58) (see Chapter 28).
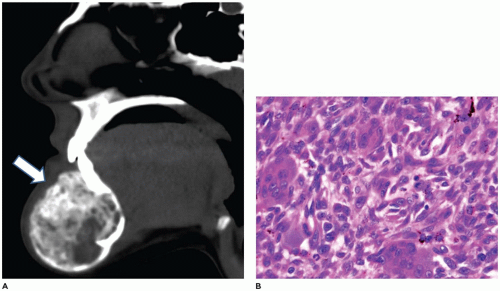 FIGURE 22-11 • Giant cell lesion of jaw. A: Anterior mandible markedly expansile lesion (arrow) on CT scan. B: Numerous osteoclast-like giant cells with variable numbers of nuclei and cellular stroma with mononuclear cells. Note the nuclear features of the giant cells and mononuclear cells are similar. |
These jaw tumors are typically treated with aggressive curettage and less often with formal resections (54,55,56,57,58). Soft tissue lesions not involving bone are conservatively excised. Treatment for aggressive tumors may involve intralesional administration of corticosteroids, interferon-á-2A, or subcutaneous or nasal spray calcitonin. Recurrence rates vary from 10% to 35%. Curettage of recurrent tumors is usually performed with good outcomes. Aggressive and/or recurrent tumors tend to possess large giant cells that are evenly distributed throughout the lesion. Prior to surgical treatment, the patients should be evaluated for cherubism, hyperparathyroidism, and the solid variant of aneurysmal bone cyst, which may have similar giant cell lesions involving the jaws.
CRANIOFACIAL OSTEOMA
Craniofacial osteomas (CFO) are typically found incidentally and are asymptomatic (60,61,62,63,64,65,66). With larger CFOs, the tumors may present as facial swelling or asymmetry. Sinus obstruction may also occur with mucocele formation and sinus discharge. When involving the orbital bones, there may be exophthalmos and inflammation of the tear duct (dacryocystitis). CFOs typically develop on the surface of the craniofacial bones and tend to more commonly involve the frontal and paranasal sinuses and the orbit. Although the tumor may arise on the surface of the frontal or paranasal sinus, the tumor may involve and obliterate the sinus (Figure 22-12). These tumors vary considerably in maximum dimension but are typically 0.5 to 2 cm in diameter. The osteoma is comprised of dense bone that merges with cortical
bone. With the craniofacial region, surgical removal consists of curettage with fragmentation of the lesion, making it difficult to determine the cortical surface from the underlying lesional tissue. Microscopic examination of the fragmented specimen demonstrates an admixture of lamellar and woven bone (Figure 22-12). The cancellous component shows intersecting broad trabeculae of bone. It is not unusual to note a more cellular component resembling osteoblastoma or a nidus usually associated with osteoid osteoma. Osteomas tend to be slow growing. Osteomas in a young individual necessitate elimination of Gardner syndrome from consideration. This can be done based upon clinical findings and/or constitutional genetic testing for APC gene mutations. Treatment of CFOs is surgical with curettage being the most common procedure with close follow-up for evaluating recurrence. There is no evidence that malignant transformation occurs (see Chapter 28).
bone. With the craniofacial region, surgical removal consists of curettage with fragmentation of the lesion, making it difficult to determine the cortical surface from the underlying lesional tissue. Microscopic examination of the fragmented specimen demonstrates an admixture of lamellar and woven bone (Figure 22-12). The cancellous component shows intersecting broad trabeculae of bone. It is not unusual to note a more cellular component resembling osteoblastoma or a nidus usually associated with osteoid osteoma. Osteomas tend to be slow growing. Osteomas in a young individual necessitate elimination of Gardner syndrome from consideration. This can be done based upon clinical findings and/or constitutional genetic testing for APC gene mutations. Treatment of CFOs is surgical with curettage being the most common procedure with close follow-up for evaluating recurrence. There is no evidence that malignant transformation occurs (see Chapter 28).
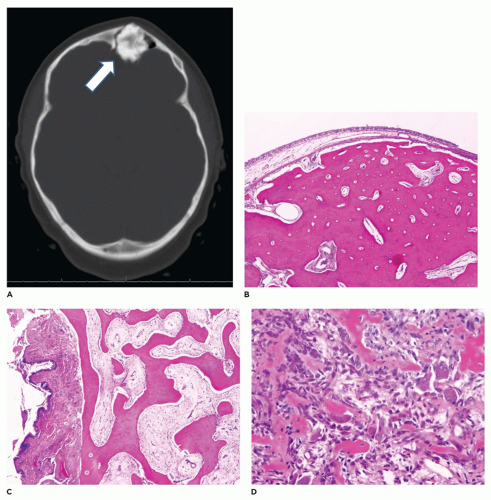 FIGURE 22-12 • Craniofacial osteoma. A: Expansile bone-forming lesion involving frontal bone (arrow) on CT scan. B: Respiratory mucosa from frontal sinus overlies dense cortical bone. C: Thin cortex and trabecular bone with fibrotic marrow from frontal sinus lesion. D: Cellular area resembling nidus for osteoid osteoma or osteoblastoma may occur with this entity. |
JUVENILE OSSIFYING FIBROMA (JUVENILE ACTIVE/AGGRESSIVE OSSIFYING FIBROMA)
Juvenile ossifying fibroma (JOF) is divided into two distinct entities: trabecular JOF (TJOF) and psammomatoid JOF (PJOF) (66,67,68,69,70,71,72,73). The majority of TJOF affects children and adolescents (mean age ranges from 8.5 to 12 years), with only 20% of affected individuals over 15 years of age. Both genders are equally affected. The maxilla and mandible (gnathic bones) are most commonly involved, with other extragnathic craniofacial bones representing a minority of lesions. Although pain is rarely associated with TJOF, the lesions tend to be progressive and may rapidly expand. With maxillary involvement, there may be epistaxis and nasal obstruction. Diagnostic imaging identifies a relatively well-demarcated expansile lesion (Figure 22-13) that may show cortical thinning and even cortex perforation. The lesion may be radiolucent or radiodense depending upon the degree of calcification. Trabecular JOF is comprised of a markedly cellular stroma with immature osteoid trabeculae without obvious osteoblasts or osteoclast rimming (Figure 22-13). The stroma contains spindled to polygonal cells with a paucity of collagen. Typical mitotic figures may be seen and these tend to be sparse. The immature osteoid trabeculae vary from long and slender to broad. Irregular, disorderly calcifications may be seen at the center of the trabeculae. Lamellar bone is not appreciated. Cystic changes may be seen in infrequent cases. Conservative excision or curettage is the treatment. Recurrence is not unusual due to the inability to perform a complete resection without considerable morbidity in most cases. Malignant transformation does not occur.
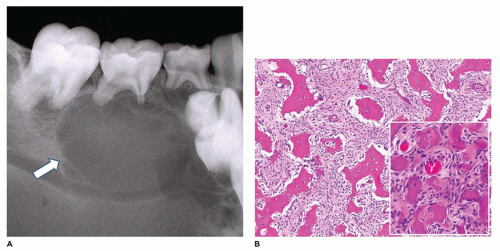 FIGURE 22-13 • Juvenile ossifying fibroma. A: Relatively well-defined radiolucent lesion (arrow) in mandible with tooth root resorption of primary teeth and displacement of developing permanent teeth on panoramic radiograph. B: Cellular trabecular pattern. Inset: Psammomatoid pattern. |
In contrast to TJOF, PJOF involves primarily extragnathic cranial bones, such as those in periorbital, frontal, and ethmoid regions, and less frequently the calvarium, maxilla, and mandible (66,67,68,69,70,71,72,73). Affected individuals tend to be young, but the age range spans from 3 months to 72 years. There is no gender predilection. These tumors present as expansile lesions. With orbital tumors, there may be proptosis, blindness, nasal obstruction, ptosis, and papilledema. Diagnostic imaging tends to show an ovoid, well-demarcated osteolytic lesion with a cystic character.
The tumor varies in size from 2 to 8 cm. Gross examination of the lesional tissue demonstrates yellow-white and gritty tissue. Microscopic examination identifies numerous round, ossicle-like, psammomatoid bodies embedded in a cellular stroma (Figure 22-13). The psammomatoid bodies resemble dental cementum. At the periphery of the lesion, the psammomatoid bodies tend to fuse to form irregular thin trabeculae. The stroma is composed of bland spindle cells and matrix that lacks a collagenous background. Of note, cystic degeneration and aneurysmal bone cyst-like areas have been described with some lesions. Treatment is surgical excision or curettage. Recurrence rates are variable and depend upon whether complete eradication of the lesional tissue is accomplished. There is no evidence of malignant transformation. Of note, cytogenetic studies have been performed in a minority of PJOF cases. However, a translocation involving Xq26 and 2q33 has been identified (see Chapter 28).
The tumor varies in size from 2 to 8 cm. Gross examination of the lesional tissue demonstrates yellow-white and gritty tissue. Microscopic examination identifies numerous round, ossicle-like, psammomatoid bodies embedded in a cellular stroma (Figure 22-13). The psammomatoid bodies resemble dental cementum. At the periphery of the lesion, the psammomatoid bodies tend to fuse to form irregular thin trabeculae. The stroma is composed of bland spindle cells and matrix that lacks a collagenous background. Of note, cystic degeneration and aneurysmal bone cyst-like areas have been described with some lesions. Treatment is surgical excision or curettage. Recurrence rates are variable and depend upon whether complete eradication of the lesional tissue is accomplished. There is no evidence of malignant transformation. Of note, cytogenetic studies have been performed in a minority of PJOF cases. However, a translocation involving Xq26 and 2q33 has been identified (see Chapter 28).
PERIPHERAL OSSIFYING FIBROMA (CEMENTO-OSSIFYING FIBROMA) OF THE ORAL CAVITY
This benign reactive proliferation is derived from pluripotential cells of the periodontal ligament and periosteum that attach the cementum root surface to the alveolar bone (74,75,76,77,78,79). These pluripotential mesenchymal cells have the ability to undergo differentiation to osteoblasts, cementoblasts, and fibroblasts. This differentiation is a reactive process to inflammation and not considered to be a true neoplastic process, such as the ossifying fibroma involving the bony structures of the head and neck region. Peripheral ossifying fibroma (POF) occurs in the submucosa subjacent to gingival tissue. This reactive lesion is typically a 1- to 2-cm lobulated painless mass that is covered by gingiva, which may undergo erosion secondary to trauma and may be associated with long-standing calculus and plaque accumulation, poor oral hygiene, orthodontic appliances, and ill-fitting dentures. The lesion may resemble a traumatic or irritation fibroma; however, POF contains spicules of bone or cementum. Adolescents and young adults are most commonly affected. Females are more commonly affected than males. Fine scattered calcifications and opacities may be seen on routine radiographic examination. Microscopic examination may be somewhat variable with some POFs showing a submucosal cellular proliferation of ovoid to spindled mesenchymal cells or paucicellular fibrotic tissue with occasional typical fibroblasts (Figure 22-14). The hallmark of this lesion that differentiates it from a traumatic or irritation fibroma is the presence of dystrophic calcifications and/or spicules and trabecula of bone and/or cementum. The bone may have either a woven or lamellar pattern. The overlying gingiva may show reactive features, such as areas of erosion and submucosal inflammation. The lesion tends to be relatively well demarcated. Treatment
involves surgical excision, deep surgical site curettage, as well as root planning of the associated teeth.
involves surgical excision, deep surgical site curettage, as well as root planning of the associated teeth.
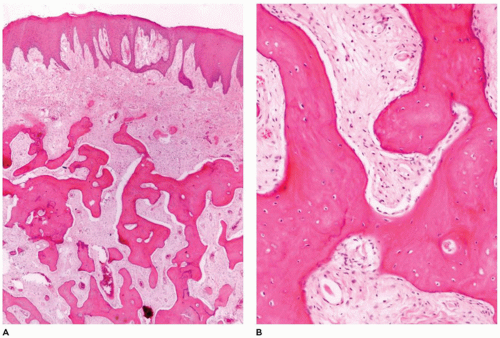 FIGURE 22-14 • Peripheral ossifying fibroma. A and B: Gingiva with intact stratified squamous epithelium (A) and underlying submucosa with bony trabeculae (A, B) with a background of low-cellularity fibrous tissue. |
DESMOPLASTIC FIBROMA
Desmoplastic fibroma (juvenile aggressive fibromatosis) is a benign intraosseous fibroblastic proliferation that is locally infiltrative and aggressive (80,81,82,83,84). This lesion occurs primarily in children and young adults, with 85% of cases occurring before age 30 years and a mean age of 14 years. Typically, the tumor involves the body-ramus of the mandible, less often the maxilla, and often erodes the bone and extends into the soft tissues (Figure 22-15). The size of the tumor varies from 3 to 9 cm and often has a lobulated soft tissue surface, and the cut surface has a firm tan-white fibrous character (Figure 22-15). The tumor is comprised of spindle cells with varying cellularity and may have a somewhat fascicular pattern with interspersed areas of collagen (Figure 22-15). Cytologic atypia, mitoses, and bone formation are absent within this tumor. The cellularity tends to be greatest at the periphery of the tumor. In contrast to desmoid tumor associated with Gardner syndrome and familial adenomatous polyposis syndrome, nuclear beta-catenin expression on immunohistochemistry is rare. The etiology of desmoplastic fibroma is unknown; however, it has been suggested that this lesion may be an exuberant reactive process. Treatment consists of complete excision with negative surgical margins. Recurrence is about 5% with complete excision and about 30% with curettage only (see Chapter 28).
 FIGURE 22-15 • Desmoplastic fibroma. A: Destructive mandibular soft tissue lesion (arrow) with expansion into the adjacent soft tissues on CT scan. B: Gross appearance of soft tissue mass with firm cut surface. C: Low to moderately cellular spindle cell tumor with interspersed collagen deposition and no cytologic atypia.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|