gland (1). An ependymal lining accompanied by reactiveappearing astrocytes can be seen (eFigure 21-2). Typically cases show a wall that exhibits features of piloid gliosis. Approximately 5% of children with hereditary retinoblastomas have pineal cyst (10). Cyst formation is also seen in pineal neoplasms (6,11).
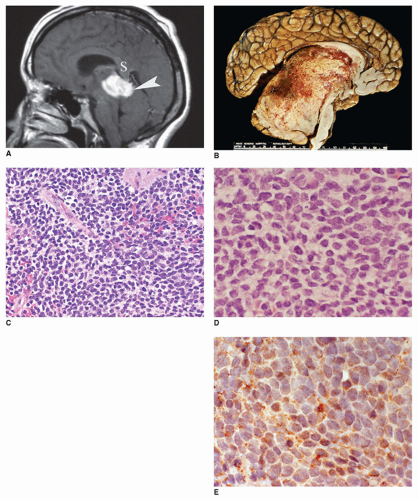 FIGURE 21-1 • Pineoblastoma in a 3-year-old girl. A: Sagittal postgadolinium T1-weighted image shows a markedly enhancing, lobulated mass (arrowhead) in the pineal region below the splenium of the corpus callosum (S). B: This large, tan-gray, infiltrative pineal tumor has a heterogeneous appearance with hemorrhage, necrosis, and leptomeningeal extension. (Used with permission, Dr. David Louis, Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts.) C: This pineal tumor is composed of sheets of primitive round to slightly ovoid cells (H&E stain, original magnification 200×). (Courtesy of Dr. Joe Parisi, Mayo Clinic, Rochester, Minnesota.) D: The tumor cells have irregular, hyperchromatic nuclei and scant cytoplasm. Mitotic figures were also present (H&E stain, original magnification 400×). (Courtesy of Dr. Joe Parisi, Mayo Clinic, Rochester, Minnesota.) E: The tumor cells demonstrate immunoreactivity with SYN (immunostain for SYN, original magnification 400×). (Courtesy of Dr. Joe Parisi, Mayo Clinic, Rochester, Minnesota.) |
during adolescence to its adult weight of 500 to 600 mg. The adenohypophysis accounts for 80% of the gland (26,27). The neonatal pituitary gland is especially prominent, owing to its stimulation by maternal hormones, but it undergoes some involution in the postnatal period, followed by increased growth through the age of 3 years. A notable increase in the size of the gland occurs with menarche and pregnancy. Generally, the pituitary gland in women after puberty weighs more than the gland in men. Suprasellar extension of the pituitary gland during puberty has been reported as a normal variant.
TABLE 21-1 CONGENITAL AND DEVELOPMENTAL ANOMALIES OF THE PITUITARY GLAND | ||||||
|---|---|---|---|---|---|---|
| ||||||
Rathke pouch in the roof of the oronasopharynx, the source of the pharyngeal pituitary gland, has been reported in a number of conditions, including the anencephalic fetus, spina bifida, trisomy 18, and Meckel syndrome. Ectopia of the posterior pituitary has been associated with mutations in genes responsible for pituitary organogenesis (eFigure 21-18) (43). Ectopic pituitary adenomas (PAs) are documented in the suprasellar region, clivus, nasopharynx, and paranasal sinuses mainly in adults, but also in children.
TABLE 21-2 MANIFESTATIONS ASSOCIATED WITH MUTATIONS IN SELECTED GENES INVOLVED IN PITUITARY DEVELOPMENT | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
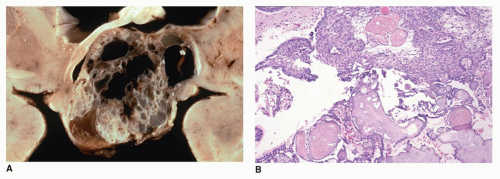 FIGURE 21-2 • Craniopharyngioma. A: This gross brain image shows a suprasellar cystic lesion filled with a dark brown fluid containing cholesterol debris. B: This adamantinomatous variant consists of ribbons of epithelial cells with pseudopalisaded nuclei at the periphery of the lobules surrounding cystic spaces. The inner cells in the more solid areas have a loose, stellate appearance. The so-called wet keratin is seen as intermixed stacks of necrobiotic squames. This image is from a 7-year-old girl, who presented with headaches and decreased visual acuity and was found to have a suprasellar mass (H&E stain). |
of the lactotrophs (responsible for prolactin secretion) and decreases in size postpartum (27).
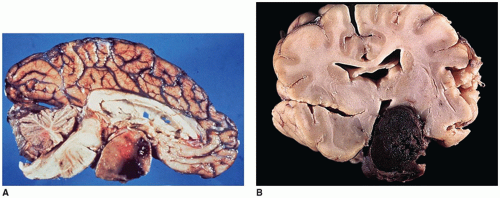 FIGURE 21-3 • Pituitary apoplexy. A: Sagittal section of brain showing hemorrhage within a pituitary macroadenoma. B: Coronal section showing hemorrhagic infarction of a 2-cm diameter well-circumscribed pituitary macroadenoma. |
and immunohistochemistry are adjuncts to characterize these tumors (Figure 21-4E, eFigure 21-33). In addition to specific hormonal immunostaining, PAs are positive for SYN, CHR, and NSE (30).
TABLE 21-3 ENDOCRINE ORGANS INVOLVED IN SELECTED FAMILIAL TUMOR SYNDROMES | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
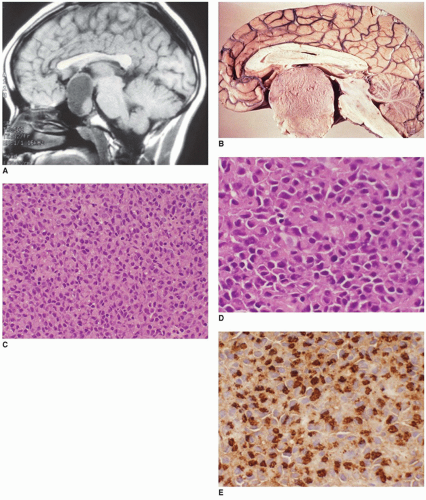 FIGURE 21-4 • Pituitary adenoma. A: A PA is shown in this sagittal T1W MR image of an 11-year-old boy with a cystic expansile mass (macroadenoma) arising within the sella turcica and extending upward (Courtesy of James Smirniotopoulos, M.D., Bethesda, Maryland). B: Sagittal section of brain showing a pituitary macroadenoma, prolactinoma, with a homogeneous cut surface. C: The normal architecture of the pituitary gland is replaced by a diffuse growth pattern of cells. The normal histologic pattern of acidophils, basophils, and chromophobes arranged in a cord-like pattern is replaced by a single population of cells with acidophilic cytoplasm (H&E stain, original magnification 200×). D: The tumor cells are large with irregular nuclei and acidophilic cytoplasm (H&E stain, original magnification 400×). E: Tumor cells are immunoreactive for prolactin in this pituitary macroadenoma (Immunostain for prolactin, original magnification 400×). (Images C-E, courtesy of Dr. Joe Parisi, Mayo Clinic, Rochester, Minnesota.) |
parathyroid glands separate and the inferior parathyroid glands localize to the inferior aspect of the thyroid gland. Formation of the parathyroid glands is associated with the genes EOLVO and GCM2 on chromosome 6 (6p24.2). Other genes associated with parathyroid development and migration include HOX3a (12q13), PAX1 (7q36), EYA1 (8q13.3, branchiootorenal syndrome 1), SIX1 (14q23, branchiootorenal syndrome), and TBX1 (22q11.2, DiGeorge syndrome). Dysregulation or mutation in these genes results in absence, hypoplasia, or ectopic parathyroid glands.
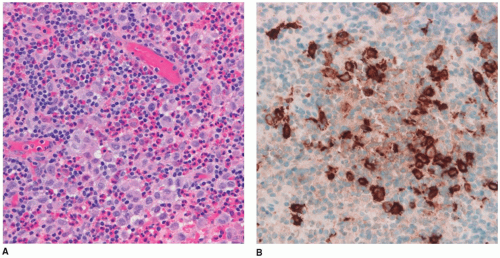 FIGURE 21-5 • Langerhans cell histiocytosis. A: Biopsy from the pituitary stalk in an adolescent with diabetes insipidus. The infiltrate is composed of foamy histiocytes that were immunoreactive with CD1a, in a background of lymphocytes, eosinophils, and plasma cells (H&E stain, original magnification, 400×). (Courtesy of Dr. Joe Parisi, Mayo Clinic, Rochester, Minnesota.) B: CD1a positivity in histiocytic cells in a patient with LCH (immunostain for CD1a, original magnification 400×). (Courtesy of Dr. Joe Parisi, Mayo Clinic, Rochester, Minnesota.) |
scintigraphic studies and US provides the highest accuracy for preoperative localization of hyperfunctioning glands.
TABLE 21-4 PRIMARY HYPERPARATHYROIDISM ETIOLOGY IN CHILDHOOD | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
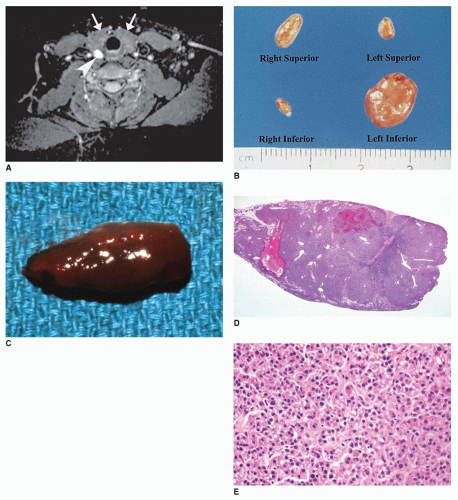 FIGURE 21-6 • Parathyroid adenoma. A: Axial T2-weighted MR image shows the hyperintense parathyroid adenoma (arrowhead) posterior to the thyroid gland (arrows) in a 14-year-old girl. B: Parathyroid adenoma in a child with primary HPT is seen as a solitary enlargement of the left inferior gland. (Courtesy of Robert Dufour, M.D., Washington, DC.) C: Parathyroid adenoma in a 16-year-old girl who presented with flank pain due to nephrolithiasis, elevated serum calcium, decreased phosphate, and increased parathormone levels. The parathyroid gland was enlarged and red-brown on gross examination. D: The enlarged parathyroid gland shows a hypercellular parenchyma composed of chief cells without intraglandular fat on low power. Necrosis was absent (H&E stain). E: The parathyroid gland is composed of a monotonous population of chief cells with no intraglandular fat. Mitotic figures were absent (H&E stain). |
attached to adjacent thyroid tissue and surrounding soft tissue. Microscopic examination shows a fibrous to fibromembranous cyst wall with parathyroid tissue embedded within the cyst wall. Only rarely have parathyroid cysts been associated with MEN syndromes.
with an intraoperative PTH level, which normalizes within a few minutes in the case of a single gland adenoma, whereas it initially falls and returns to an elevated level in the presence of hyperplasia (74). An enlarged gland may be occult within the thymus, thyroid, or paraesophageal. Ultrasound and nuclear medicine scans are important in localizing abnormal glands prior to surgery.
familial (non-MEN) MTC (FMTC) is the consequence of germline RET gene mutations (Table 21-2). These syndromes are characterized by multifocal C-cell hyperplasia and often multifocal MTCs (84,86,87).
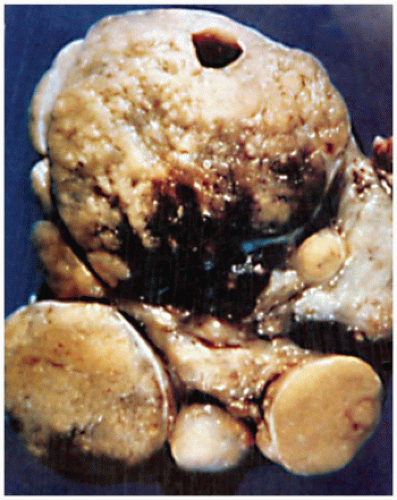 FIGURE 21-7 • Dyshormonogenic goiter. This image is a section through the thyroid gland of an individual who presented with a dyshormonogenic goiter. The thyroid parenchyma has a nodular pattern with retrogressive and hyperplastic changes including hemorrhage and fibrosis. (From Lloyd RV, Douglas BR, Young WF. Endocrine diseases. Atlas of Nontumor Pathology. Washington, DC: American Registry of Pathology, 2001. Originally published in Atlas of tumor pathology, tumors of the thyroid gland, Fascicle 5, Third Series. Washington, DC: Armed Forces Institute of Pathology). |
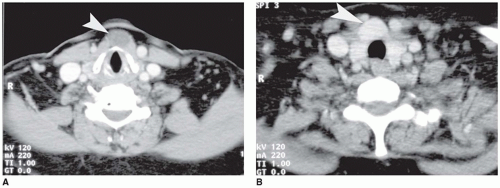 FIGURE 21-8 • Thyroglossal duct cyst in an adult complicated by PTC. A: Axial contrastenhanced CT image shows a midline cyst (arrowhead). B: Axial CT image caudal to (a) shows markedly enhancing, midline mass (arrowhead). |
of thyroid tissue (hypoplasia), absence of a lobe (hemiagenesis), or ectopic location. Dysgenesis is an important etiology of congenital hypothyroidism, with prevalence in the United States of 1:3000 to 5000 live births. Most causes of congenital hypothyroidism are due to dysgenesis or inherited defects in thyroid hormone synthesis (Table 21-5). Congenital hypothyroidism has been also been observed in Williams and Down syndromes.
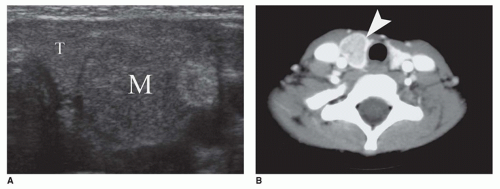 FIGURE 21-9 • Papillary thyroid carcinoma in 15-year-old girl. A: Transverse sonographic image showing a heterogeneous mass (M) within the homogeneous thyroid gland (T). B: MTC in an 8-year-old girl with family history of MEN2a. Axial contrast-enhanced CT shows a mass within the left thyroid lobe, which enhances less than the surrounding thyroid gland (arrowhead). |
gland is anatomically orthotopic in the presence of congenital hypothyroidism, a defect exists in thyroid hormone biosynthesis with the development of a dyshormonogenic nodular goiter or an inability of the gland to respond to TSH. Dysgenesis is more common in females than in males (3:1) and is sporadic in most cases (85%) (79). Affected infants with agenesis or hypoplasia have permanently elevated levels of TSH and low levels of circulating thyroid hormone. A number of mutations have been identified in the genes responsible for thyroid development, including PAX8, TTF-1 (thyroid transcription factor-1), TTF-2 (thyroid transcription factor-2), TSHR (TSH receptor), thyroglobulin (8q24), thyroperoxidase (2p25), sodium-iodide symporter (19p13), GNAS 1 (20q13), pendrin (7q31), DUOX2 (15q15), and DUOXA2 (15q15) and DHAL1 (6q24) and are pathogenetically involved in thyroid dysgenesis (79,80,81,82,83,92). These genetic defects and their association with other diseases are reviewed elsewhere (79).
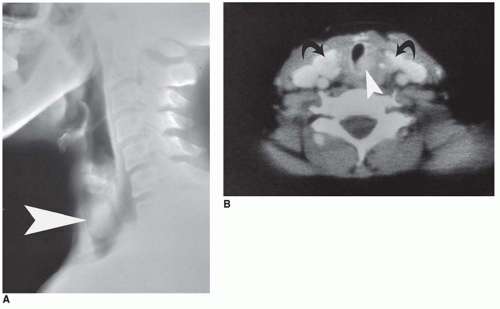 FIGURE 21-10 • Ectopic thyroid gland in the trachea of an adult. A: Lateral tomogram shows an ovoid mass within the tracheal air column (arrowhead). B: Axial CT image shows markedly enhancing eccentric mass in the trachea (arrowhead) and normal thyroid lobes in the orthotopic location (curved arrows). |
TABLE 21-5 ETIOLOGIC CLASSIFICATION OF CONGENITAL HYPOTHYROIDISM | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
laterally, usually on the left side, and a minority occurs at the base of the tongue, floor of the mouth, or within the thyroid itself. The TDC differential includes branchial cleft cyst (lymphoepithelial cyst), lymphangioma, lymphadenopathy, lymphoma, epidermal inclusion cyst, and other thyroid malformations (94). Most cysts are diagnosed at or before 5 years of age as a painless cystic midline cervical mass, but may be recognized throughout life (56,94). Because of possible communication with the oral cavity through the foramen cecum, TDCs may become infected, and there may be periodic oral drainage with halitosis. Cutaneous draining sinuses may also occur in the midline of the neck through a fistulous or sinus tract. A familial association has been reported. A rare presentation of TDC is sudden death due to asphyxiation (56). Infected cysts may lead to fistula formation to the skin surface or pharynx (56).
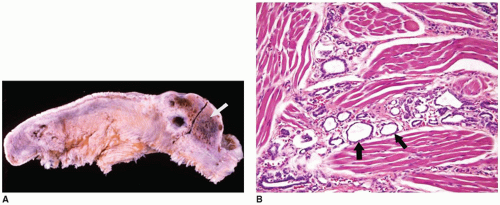 FIGURE 21-11 • Lingual thyroid. A: Sagittal section through the tongue, which shows a smooth, ovoid, 2-cm-diameter mass in the posterior third of the tongue (arrow). Small and large cysts with adjacent red-brown thyroid tissue are present in the mass. Incidental finding at autopsy in a 69-year-old man who died from cerebral hemorrhage. (From Turk JL, Fletcher CDM, eds. Endocrine System. Royal College of Surgeons of England Slide Atlas of Pathology, 1985. Originally published by Gower Medical Publishing, Ltd. Reprinted with permission of Elsevier Inc. and CDM Fletcher, M.D.). B: This section of tongue shows the presence of thyroid follicles between the muscle fibers (arrows). This was an incidental finding at autopsy in a stillborn infant (H&E stain, original magnification 100×). |
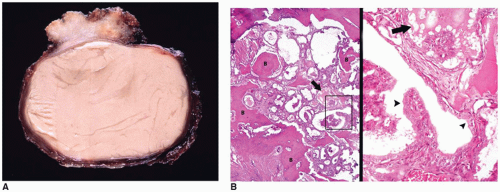 FIGURE 21-12 • Thyroglossal duct cyst. A: This midline cyst was filled with tan-white mucoid fluid on gross examination. Fibrosis of soft tissue adjacent to the cyst was present. B: This composite image shows the resected hyoid bone on the left with entrapped thyroid follicles (arrow). The area within the rectangle is magnified on the right side and shows a cuboidal epithelium (arrowheads) lining the cystic spaces. A thyroid follicle is also present in this image (arrow). Lymphoid aggregates not shown were also present (H&E stain). |
autoimmune diathesis . Approximately 4% of children with type I diabetes mellitus have CLT. Trisomy 21 syndrome, Klinefelter syndrome, and Turner syndrome are three chromosomal disorders associated with CLT. Approximately 25% of young individuals with Turner syndrome have antithyroid antibodies and 10% have enlarged thyroids.
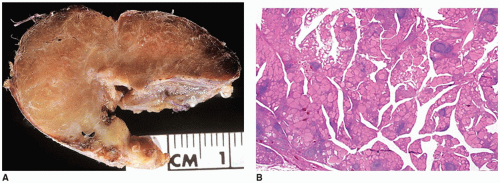 FIGURE 21-13 • Chronic lymphocytic thyroiditis. A: This specimen shows the characteristic diffuse thyroid gland enlargement seen in CLT on gross examination. A vaguely nodular pattern corresponding to the presence of lymphoid follicles is seen in this cut section. B: CLT in this low-power magnification image shows prominent lymphoid aggregates interspersed between the thyroid follicles. Plasma cells and lymphocytes were present in the interstitium (H&E stain). |
thyroid gland without obvious evidence of hyperthyroidism. Children with a simple goiter are predominantly young adolescent females and do not experience any further gland enlargement. A small percentage, however, may develop CLT.
infoldings, is seen in the gland treated by thiouracil. Iodine administration before surgery results in the accumulation of colloid and the formation of macrofollicles, often with associated accentuation of thyroid lobules by thin fibrous bands. Rather than cuboidal to columnar epithelium, flattened epithelial cells cover the slender papillae.
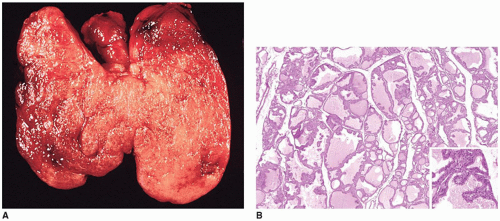 FIGURE 21-14 • Graves disease. A: This image shows diffuse symmetrical enlargement of the thyroid gland from a patient with Graves disease. The parenchyma has a deep red color due to increased vascularity within the gland. (Reprinted with permission from Lloyd RV, Douglas BR, Young WF. Endocrine Diseases. Atlas of Nontumor Pathology. Washington, DC: American Registry of Pathology, 2001.) B: This section of thyroid gland from a patient with untreated Graves disease shows follicles with hyperplastic epithelium and papillary infoldings. Pale watery colloid and an interstitial lymphocytic infiltrate (not pictured) were observed. The papillary infoldings (inset) lack the optically clear nuclei seen in PTC (H&E stain). |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


