68
Immunodeficiency
CHAPTER CONTENTS
INTRODUCTION
Immunodeficiency can occur in any of the four major components of the immune system: (1) B cells (antibody), (2) T cells, (3) complement, and (4) phagocytes. The deficiencies can be either congenital or acquired (Table 68–1). Clinically, recurrent or opportunistic infections are commonly seen. Recurrent infections with pyogenic bacteria (e.g., staphylococci) indicate a B-cell deficiency, whereas recurrent infections with certain fungi, viruses, or protozoa indicate a T-cell deficiency.
TABLE 68–1 Important Congenital Immunodeficiencies
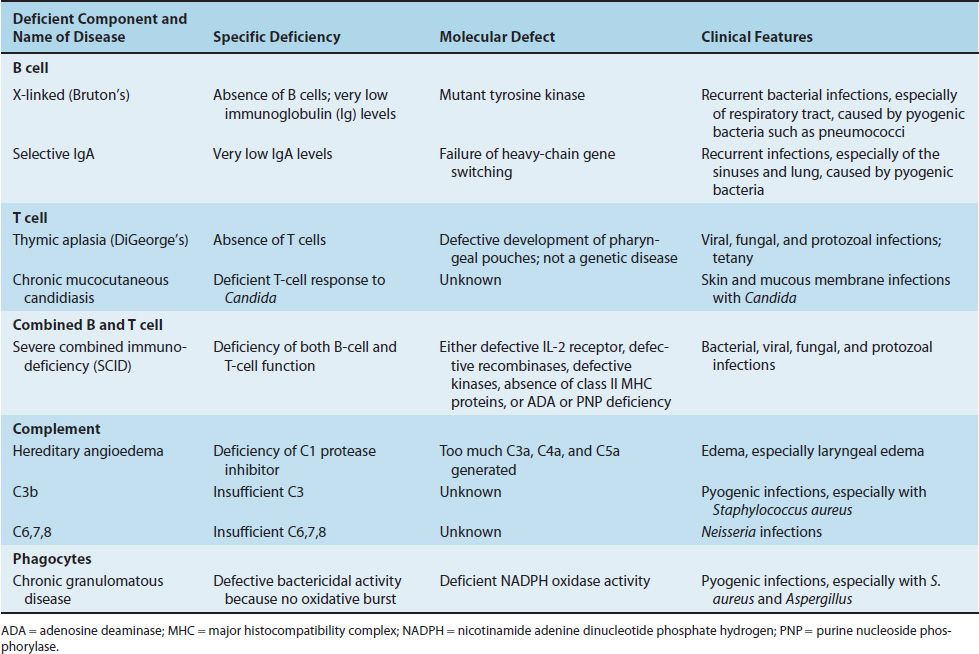
CONGENITAL IMMUNODEFICIENCIES
B-Cell Deficiencies
X-Linked Hypogammaglobulinemia (Bruton’s Agammaglobulinemia)
Very low levels of all immunoglobulins (IgG, IgA, IgM, IgD, and IgE) and a virtual absence of B cells are found in young boys; female carriers are immunologically normal. Pre-B cells are present, but they fail to differentiate into B cells. This failure is caused by a mutation in the gene encoding tyrosine kinase, an important signal transduction protein. Cell-mediated immunity is relatively normal. Clinically, recurrent pyogenic bacterial infections (e.g., otitis media, sinusitis, and pneumonia caused by Streptococcus pneumoniae and Haemophilus influenzae) occur in infants at about 6 months of age, when maternal antibody is no longer present in sufficient amount to be protective. Treatment with pooled gamma globulin reduces the number of infections.
Selective Immunoglobulin Deficiencies
IgA deficiency is the most common of these; IgG and IgM deficiencies are rarer. Patients with a deficiency of IgA typically have recurrent sinus and lung infections. (However, some individuals with IgA deficiency do not have frequent infections, possibly because their IgG and IgM levels confer protection.) The cause of IgA deficiency may be a failure of heavy chain gene switching because the amounts of IgG and IgM are normal. Patients with a deficiency of IgA should not be treated with gamma globulin preparations, because these patients may form antibodies against the foreign IgA and, by cross-reaction, deplete their already low level of IgA.
Patients with selective IgM deficiency or deficiency of one or more of the IgG subclasses also have recurrent sinopulmonary infections caused by pyogenic bacteria such as S. pneumoniae, H. influenzae, or Staphylococcus aureus.
T-Cell Deficiencies
Thymic Aplasia (DiGeorge’s Syndrome)
Severe viral, fungal, or protozoal infections occur in affected infants early in life as a result of a profound deficit of T cells. Pneumonia caused by Pneumocystis jiroveci and thrush caused by Candida albicans are two common infections in these patients. Antibody production may be decreased or normal. If decreased, severe pyogenic bacterial infections can occur.
Both the thymus and the parathyroids fail to develop properly as a result of a defect in the third and fourth pharyngeal pouches. The most common presenting symptom is tetany due to hypocalcemia caused by hypoparathyroidism. Other congenital abnormalities are common. A transplant of fetal thymus may reconstitute T-cell immunity. A thymus from a child older than 14 weeks should not be used, because a graft-versus-host reaction may occur.
Chronic Mucocutaneous Candidiasis
In this disease, the skin and mucous membranes of children are infected with C. albicans, which in immunocompetent individuals is a nonpathogenic member of the normal flora. These children have a T-cell deficiency specifically for this organism; other T-cell and B-cell functions are normal. Treatment consists primarily of antifungal drugs.
Hyper-IgM Syndrome
In this syndrome, severe, recurrent pyogenic bacterial infections resembling those seen in X-linked hypogammaglobulinemia begin early in life. Patients have a high concentration of IgM but very little IgG, IgA, and IgE. They have normal numbers of T cells and B cells. Although the main manifestations of this syndrome are alterations in antibodies, the mutation is in the gene encoding the CD40 ligand in the CD4-positive helper T cells. As a result, the helper T cells have a defect in the surface protein (CD40 ligand) that interacts with CD40 on the B-cell surface. The failure to properly interact with CD40 results in an inability of the B cell to switch from the production of IgM to the other classes of antibodies. Treatment with pooled gamma globulin results in fewer infections.
Interleukin-12 Receptor Deficiency
Patients with a deficiency of interleukin (IL)-12 receptor have disseminated mycobacterial infections. The absence of the receptor prevents IL-12 from initiating a Th-1 response, which is required to limit mycobacterial infections.
Combined B-Cell & T-Cell Deficiencies
Severe Combined Immunodeficiency Disease (SCID)
Recurrent infections caused by bacteria, viruses, fungi, and protozoa occur in early infancy (3 months of age) because both B cells and T cells are defective. In some children, the B and T cells are absent; in others, the number of cells is normal but they do not function properly. Immunoglobulin levels are very low, and tonsils and lymph nodes are absent. Pneumocystis pneumonia is the most common presenting infection in these infants. Infections caused by C. albicans and viruses such as varicella-zoster virus, cytomegalovirus, and respiratory syncytial virus are common and often fatal.
This is a group of inherited diseases, all of which are due to a defect in the differentiation of an early stem cell. There are two types: X-linked and autosomal; the X-linked form constitutes about 75% of cases. Some patients with X-linked SCID have a defect in the IL-2 receptor on T cells. They lack the γ chain of the IL-2 receptor that is essential for the development of T cells. This is the most common form of SCID in the United States. The chain of the IL-2 receptor is shared with the IL-7 receptor, so SCID can also result from a failure of IL-7 to stimulate the progression of stem cells into T cells and B cells.
Some patients with the autosomal form have a mutation in the gene encoding a tyrosine kinase called ZAP-70 that plays a role in signal transduction in T cells. Another autosomal form has mutations in the gene for a different kinase called Janus kinase 3. Other SCID patients with the autosomal form have a mutation in the RAG-1 or RAG-2 genes that encode the recombinase enzymes that catalyze the recombination of the DNA required to generate the T-cell antigen receptor and the IgM monomer on the B cell that acts as the antigen receptor.
Because immunity is so profoundly depressed, these children must be protected from exposure to microorganisms, usually by being enclosed in a plastic “bubble.” Live, attenuated viral vaccines should not be given. Bone marrow transplantation may restore immunity. It is interesting that because infants with SCID do not reject allografts, bone marrow transplants do not require immunosuppressive drugs.
Patients with a hereditary absence of adenosine deaminase (ADA) and purine nucleoside phosphorylase (PNP) can have a severe deficiency of B cells and T cells, causing SCID, although some have only mild dysfunction. The absence of these enzymes results in an accumulation of deoxyadenosine triphosphate (dATP), an inhibitor of ribonucleotide reductase, and a consequent decrease in the deoxynucleoside triphosphate precursors of DNA. This reduces the formation of B-cell and T-cell precursors in the bone marrow. Bone marrow transplantation can be helpful. Injections of ADA conjugated to polyethylene glycol reduce the number and severity of infections. Several patients with ADA deficiency have benefited from gene therapy. A retroviral vector carrying a normal copy of the ADA gene was allowed to infect the patient’s bone marrow cells. The ADA gene functioned within some of these cells, and the patient’s immune status improved.
Patients with bare lymphocyte syndrome exhibit the signs and symptoms of SCID and are especially susceptible to viral infections. These patients have defective class I or class II major histocompatibility complex (MHC) proteins or both. Mutations resulting in the inability to synthesize a transcription factor required for the synthesis of the mRNA for class II MHC proteins are an important cause of the failure to produce those proteins. Mutations in the gene encoding the TAP protein have been identified as one cause of the inability to display antigens on class I MHC proteins. (The TAP transporter protein is described on page 494.)
Wiskott-Aldrich Syndrome
Recurrent pyogenic infections, eczema, and bleeding caused by thrombocytopenia characterize this syndrome. These symptoms typically appear during the first year of life. It is an X-linked disease and thus occurs only in male infants. The most important defect is the inability to mount an IgM response to the capsular polysaccharides of bacteria, such as pneumococci. IgG levels and IgA levels are normal, but cell-mediated immunity is variable. The defect appears to be in the ability of T cells to provide help to B cells. The mutant gene encodes a protein involved in actin filament assembly. Bone marrow transplantation may be helpful.
Ataxia–Telangiectasia
In this disease, ataxia (staggering), telangiectasia (enlarged small blood vessels of the conjunctivas and skin), and recurrent infections appear by 2 years of age. It is an autosomal recessive disease caused by mutations in the genes that encode DNA repair enzymes. Lymphopenia and IgA deficiency commonly occur. Treatment designed to correct the immunodeficiency has not been successful.
Complement Deficiencies
Hereditary Angioedema
This is an uncommon autosomal dominant disease caused by a deficiency of C1 inhibitor. In the absence of inhibitor, C1 continues to act on C4 to generate C4a and subsequently additional vasoactive components such as C3a and C5a. This leads to capillary permeability and edema in several organs. Laryngeal edema can be fatal. Steroid drugs, such as oxymetholone and danazol, can be useful in increasing the concentration of C1 inhibitor.
Recurrent Infections
Patients with deficiencies in C1, C3, or C5 or the later components C6, C7, or C8 have an increased susceptibility to bacterial infections. Patients with C3 deficiency are particularly susceptible to sepsis with pyogenic bacteria such as S. aureus. Those with reduced levels of C6, C7, or C8 are especially prone to bacteremia with Neisseria meningitidis or Neisseria gonorrhoeae.
Autoimmune Diseases
Patients with C2 and C4 deficiencies have diseases resembling systemic lupus erythematosus or other autoimmune diseases. C2 deficiency is the most common complement defect and is frequently asymptomatic.
Paroxysmal Nocturnal Hemoglobinuria
This rare disease is characterized by episodes of brownish urine (hemoglobinuria), particularly upon arising. The hemoglobinuria is due to complement-mediated hemolysis. This occurs especially at night because the lower oxygen concentration (and low pH) in the blood during sleep increases the susceptibility of the red cells to lyse. Hemolysis occurs because there is a reduced amount of decay-accelerating factor (DAF) bound to the surface of red blood cells, leading to an increased activation of complement (see Chapter 63). These patients have a defect in the gene for the molecules that anchor DAF and other proteins to the cell membrane. There is no specific treatment. Iron can be given for the anemia, and prednisone can be helpful.
Phagocyte Deficiencies
Chronic Granulomatous Disease (CGD)
Patients with this disease are very susceptible to opportunistic infections with certain bacteria and fungi (e.g., S. aureus); enteric gram-negative rods, especially Serratia and Burkholderia; and Aspergillus fumigatus. Recurrent infections with catalase-positive bacteria, such as staphylococci, are common in these patients, whereas infections with catalase-negative bacteria, such as streptococci, are rare. Viral, mycobacterial, and protozoal infections are not a major concern. In 60% to 80% of cases, this is an X-linked disease that appears by the age of 2 years. (In the remaining patients, the disease is autosomal.)
CGD is due to a defect in the intracellular microbicidal activity of neutrophils as a result of a lack of NADPH oxidase activity (or similar enzymes). As a result, no hydrogen peroxide or superoxides are produced (i.e., no oxidative burst occurs), and the organisms, although ingested, are not killed. B-cell and T-cell functions are usually normal.
In the laboratory, diagnosis can be confirmed by the nitroblue tetrazolium dye reduction test or by the dichlorofluorescein (DCF) test. The DCF test is the more informative of the two because the analysis is done by flow cytometry, which provides information regarding the oxidative ability of single cells. For example, in the mothers of boys with CGD who are carriers, half of their neutrophils show normal oxidative activity because the X chromosome carrying the mutant gene has been inactivated, whereas the other half show no oxidative activity because the X chromosome carrying the normal gene has been inactivated.
Prompt, aggressive treatment of infection with the appropriate antibiotics is important. Chemoprophylaxis using trimethoprim-sulfamethoxazole can reduce the number of infections. Gamma interferon significantly reduces the frequency of recurrent infections, probably because it increases phagocytosis by macrophages.
The name chronic granulomatous disease arises from the widespread granulomas seen in these patients, even in the absence of clinically apparent infection. These granulomas can become large enough to cause obstruction of the stomach, esophagus, or bladder. The cause of these granulomas is unknown.
Chédiak-Higashi Syndrome
In this autosomal recessive disease, recurrent pyogenic infections, caused primarily by staphylococci and streptococci, occur. This is due to the failure of the lysosomes of neutrophils to fuse with phagosomes. The degradative enzymes in the lysosomes are, therefore, not available to kill the ingested organisms. Large granular inclusions composed of abnormal lysosomes are seen. In addition, the neutrophils do not function correctly during chemotaxis as a result of faulty microtubules. The mutant gene in this disease encodes a cytoplasmic protein involved in protein transport. Peroxide and superoxide formation is normal, as are B-cell and T-cell functions. Treatment involves antimicrobial drugs. There is no useful therapy for the phagocyte defect.
Job’s Syndrome (Hyper-IgE Syndrome)
Patients with this syndrome have recurrent “cold”1 staphylococcal abscesses, eczema, skeletal defects, and high levels of IgE.
The main immunologic defect is a failure to produce gamma interferon by helper T cells, which reduces the ability of macrophages to kill bacteria. This leads to an increase in Th-2 cells and, as a consequence, a high IgE level. The increased IgE causes histamine release, which blocks certain aspects of the inflammatory response, hence the cold abscesses. Histamine also inhibits neutrophil chemotaxis, another feature of this syndrome. Treatment consists of antimicrobial drugs.
Leukocyte Adhesion Deficiency Syndrome
Patients with this syndrome have severe pyogenic infections early in life because they have defective adhesion (LFA-1) proteins on the surface of their phagocytes. This is an autosomal recessive disease in which there is a mutation in the gene encoding the β chain of an integrin that mediates adhesion. As a result, neutrophils adhere poorly to endothelial cell surfaces, and phagocytosis of the bacteria is inadequate.
Cyclic Neutropenia
In this autosomal dominant disease, patients have a very low neutrophil count (less than 200/μL) for 3 to 6 days of a 21-day cycle. During the neutropenic stage, patients are susceptible to life-threatening bacterial infections, but when neutrophil counts are normal, patients are not susceptible. Mutations in the gene encoding neutrophil elastase have been identified in these patients, but it is unclear how these contribute to the cyclic nature of the disease. It is hypothesized that irregular production of granulocyte colony-stimulating factor may play a role in the cyclic aspect of the disease.
Myeloperoxidase Deficiency
Deficiency of myeloperoxidase (either reduced amount or reduced function) is quite common but has little clinical importance. Surprisingly, most patients with this deficiency do not have a significant increase in infectious diseases. Myeloperoxidase catalyzes the production of hypochlorite, an important microbicidal agent, so an increase in infections would be expected. However, other intracellular killing mechanisms are intact and must be sufficient to kill the ingested microbes.
Interferon-Gamma Receptor Deficiency
Patients with this deficiency have severe infections with atypical mycobacteria or with bacillus Calmette-Guérin (BCG), the attenuated mycobacterium in the BCG vaccine. They have a mutation in the gene encoding either the ligand-binding portion or the signal-transducing portion of the receptor for interferon-gamma. As a result, macrophages are not activated, and severe mycobacterial infections occur. Defects in the production of IL-12 or in the receptor for IL-12 cause the same clinical picture.
Pattern-Recognition Receptor Deficiency
Mutations in the genes encoding the pattern-recognition receptors (PRRs) on the surface of and within the cells of the innate immune system result in susceptibility to severe infections (Table 68–2). For more information on PRRs, see the “Innate Immunity” section in Chapter 57.
TABLE 68–2 Important Pattern-Recognition Receptor Deficiencies of Innate Immune Cells
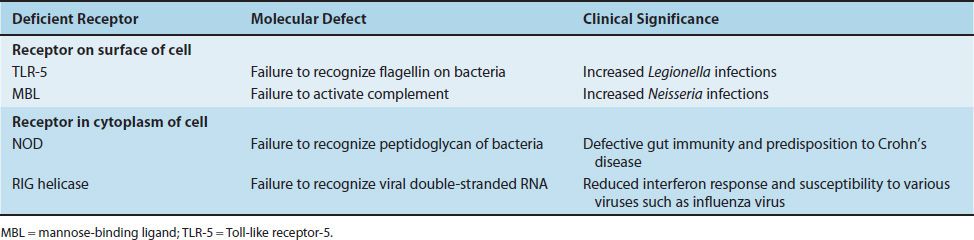
Receptors on the Surface of Innate Immune Cells
Deficiency of Toll-like receptor-5 (TLR-5) results in a failure to recognize flagellin on bacteria and a marked susceptibility to Legionella infections. This deficiency is quite common. Deficiency of mannose-binding ligand (MBL) is also common. It results in a failure to activate complement, leading to severe infections by gram-negative bacteria such as Neisseria.
Receptors Within Innate Immune Cells
NOD receptors in the cytoplasm recognize the peptidoglycan of gram-positive and gram-negative bacteria. Deficiency of NOD-2 results in a defect in gut immunity that is involved in the pathogenesis of Crohn’s disease. RIG helicase receptors recognize viral double-stranded RNAs synthesized during replication in the cytoplasm. Deficiency of these receptors results in a reduced interferon response to various viruses (i.e., influenza virus).
ACQUIRED IMMUNODEFICIENCIES
B-Cell Deficiencies
Common Variable Hypogammaglobulinemia
Patients present with recurrent infections caused by pyogenic bacteria (e.g., sinusitis and pneumonia caused by pyogenic bacteria such as S. pneumoniae and H. influenzae). The infections usually occur in persons between the ages of 15 and 35 years. The number of B cells is usually normal, but the ability to synthesize IgG (and other immunoglobulins) is greatly reduced. Cell-mediated immunity is usually normal. The cause of the failure to produce IgG is unknown but appears to be due to defective T-cell signaling. Intravenous gamma globulin given monthly reduces the number of infections.
Malnutrition
Severe malnutrition can reduce the supply of amino acids and thereby reduce the synthesis of IgG. This predisposes to infection by pyogenic bacteria.
T-Cell Deficiencies
Acquired Immunodeficiency Syndrome
Patients with acquired immunodeficiency syndrome (AIDS) present with opportunistic infections caused by certain bacteria, viruses, fungi, and protozoa (e.g., Mycobacterium avium-intracellulare, herpesviruses, C. albicans, and P. jiroveci). This is due to greatly reduced helper T-cell numbers caused by infection with the retrovirus human immunodeficiency virus (HIV; see Chapter 45). This virus specifically infects and kills cells bearing the CD4 protein as a surface receptor. The response to specific immunizations is poor; this is attributed to the loss of helper T-cell activity. AIDS patients also have a high incidence of tumors such as lymphomas, which may be the result of a failure of immune surveillance. See Chapter 45 for information on treatment and prevention.
Measles
Patients with measles have a transient suppression of delayed hypersensitivity as manifested by a loss of PPD skin test reactivity. Quiescent tuberculosis can become active. In these patients, T-cell function is altered but immunoglobulins are normal.
Complement Deficiencies
Liver Failure
Liver failure caused by alcoholic cirrhosis or by chronic hepatitis B or hepatitis C can reduce the synthesis of complement proteins by the liver to a level that severe pyogenic infections can occur.
Malnutrition
Severe malnutrition can reduce the supply of amino acids and thereby reduce the synthesis of complement proteins by the liver. This predisposes to infection by pyogenic bacteria.
Phagocyte Deficiencies
Neutropenia
Patients with neutropenia present with severe infections caused by pyogenic bacteria such as S. aureus and S. pneumoniae and enteric gram-negative rods. Neutrophil counts below 500/μL predispose to these infections. Common causes of neutropenia include cytotoxic drugs, such as those used in cancer chemotherapy; leukemia, in which the bone marrow is “crowded out” by leukemic cells; and autoimmune destruction of the neutrophils. Ciprofloxacin is used to try to prevent infections in neutropenic patients.
Chronic Fatigue Syndrome (Chronic Fatigue Immune Dysfunction Syndrome)
The predominant finding in patients with chronic fatigue syndrome (CFS) is persistent, debilitating fatigue that has lasted for at least 6 months and is not relieved by bed rest. Because fatigue is a nonspecific symptom, all other causes of fatigue, including physical (e.g., cancer, autoimmune disease, and infection) and psychiatric (e.g., depression and neurosis), as well as prolonged use of drugs (e.g., tranquilizers), must be ruled out. The cause of CFS is unknown; attempts to isolate a causative organism from these patients have failed. A proposed relationship between CFS and chronic Epstein–Barr virus infection remains unsubstantiated.
There is a similarity between the symptoms of CFS and the symptoms that occur when alpha interferon or IL-2 is administered to patients. Abnormalities in various components of the immune system have been reported (e.g., loss of delayed hypersensitivity reactivity in skin tests and increased levels of cytotoxic T cells), but no definitive findings have emerged. There is no specific laboratory test for CFS. The approach to therapy involves treating the symptoms. Treatment with various antimicrobial drugs, such as acyclovir, ketoconazole, and gamma globulin, had no effect.
SELF-ASSESSMENT QUESTIONS
1. Your patient is a 2-year-old boy who has had several episodes of pustules and lymphadenitis caused by Staphylococcus aureus. His immunoglobulins and complement levels are normal. A nitroblue tetrazolium test reveals defective cells. Which one of the following cells is the most likely to be defective?
(A) CD4-positive T lymphocytes
(B) CD8-positive T lymphocytes
(C) Eosinophils
(D) Natural killer cells
(E) Neutrophils
2. Your patient is a 25-year-old woman who has had several serious episodes of bacterial pneumonia in the last 5 months. She has not had frequent or unusual infections prior to the onset of these pneumonias. Which one of the following is the most likely immunodeficiency that predisposes her to these infections?
(A) She is likely to have a defect in her cytotoxic T cells.
(B) She is likely to have a reduced level of immunoglobulins.
(C) She is likely to have a mutation in the gene that encodes the C3a portion of complement.
(D) She is likely to have a mutation in one of the genes that encode the class I MHC proteins.
3. Regarding Bruton’s agammaglobulinemia, which one of the following is most accurate?
(A) There is very little IgG produced, but IgM and IgA levels are normal.
(B) Viral infections are more common than pyogenic bacterial infections.
(C) The number of B cells is normal, but they cannot differentiate into plasma cells.
(D) There is a mutation in the gene for tyrosine kinase, an important enzyme in the signal transduction pathway in B cells.
4. Which one of the following is the most accurate description of the defect in chronic granulomatous disease?
(A) There is an inability to produce an oxidative burst.
(B) There is a failure to produce sufficient interleukin-2.
(C) There is a deficiency of a late-acting complement component.
(D) There is a mutant protein kinase in a signal transduction pathway.
(E) There is a mutation in the gene that encodes a class II MHC protein.
5. Which one of the following immunodeficiencies is most likely to predispose to both pyogenic bacterial infections and viral infections in a young child?
(A) Bruton’s agammaglobulinemia
(B) Chronic granulomatous disease
(C) DiGeorge’s syndrome
(D) Job’s syndrome
(E) Severe combined immunodeficiency disease
6. Regarding immunodeficiency diseases, which one of the following is most accurate?
(A) Patients who have a deficiency of IgA have a high incidence of pyogenic infections of the sinuses and lungs.
(B) Common variable hypogammaglobulinemia typically occurs in boys under the age of 6 months and results from a virtual absence of B cells.
(C) In Wiskott-Aldrich syndrome, the combination of antibody deficiency and complement deficiency leads to disseminated viral and fungal infections.
(D) Patients with DiGeorge’s syndrome (congenital thymic aplasia) have a reduced number of both T cells and B cells and have severe infections caused by pyogenic bacteria.
(E) Patients who cannot produce one or more of the late-acting complement components, such as C6, C7, C8, or C9, have episodes of angioedema, including laryngeal edema that can be fatal.
ANSWERS
1. (E)
2. (B)
3. (D)
4. (A)
5. (E)
6. (A)
PRACTICE QUESTIONS: USMLE & COURSE EXAMINATIONS
Questions on the topics discussed in this chapter can be found in the Immunology section of Part XIII: USMLE (National Board) Practice Questions starting on page 713. Also see Part XIV: USMLE (National Board) Practice Examination starting on page 731.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


