Hepatitis A Virus
F. Blaine Hollinger
Annette Martin
History
Reports of icteric disease in ancient Chinese literature, as reviewed by Zuckerman,419 and a letter from Pope Zacharias to Archbishop St. Boniface of Mainz during the 8th century AD describing an outbreak of jaundice may refer to hepatitis A; however, it cannot be distinguished from jaundice attributable to other causes. It was not until the 17th, 18th, and 19th centuries that scattered epidemics of jaundice affecting diverse populations were recorded more frequently. The disease, which was common in military troops, was called campaign jaundice. In time, an infectious cause was suspected by several clinicians, especially for the milder, more contagious forms called epidemic catarrhal jaundice.143 In 1912, Cockayne54 used the term infectious hepatitis to describe this epidemic form of the disease, and support for a viral cause gained favor from that point forward.260
Epidemiologic studies and human volunteer experiments performed during and after World War II confirmed the viral cause of hepatitis A and defined its relatively short incubation period and its fecal–oral mode of transmission. These studies also demonstrated the uniqueness of this disease from homologous serum jaundice, another form of hepatitis having a longer incubation period.30,153,154,205 In 1947, MacCallum235 introduced the terms hepatitis A and hepatitis B to categorize these diseases, terms that were adopted in 1973 by the World Health Organization (WHO) Expert Committee on Viral Hepatitis.401 Between the mid-1950s and early 1970s, the studies of Murray270 and of Krugman et al205,207 at the Willowbrook State School for the mentally handicapped were instrumental in further defining the seroepidemiologic relationships between hepatitis A and B that eventually led to the evaluation of new methods for the immunoprophylaxis of these diseases. One of the hepatitis viruses, designated MS-1, was transmitted primarily by the fecal–oral route. The disease was highly infectious, had a relatively short incubation period of 4 ± 2 weeks, and defined classic viral hepatitis type A. The MS-1 strain of hepatitis A virus (HAV) was later used to infect adult volunteers,30 leading to the identification of the virus in their feces by immune electron microscopy, as reported by Feinstone et al101 in 1973. Highly sensitive immunoassays were subsequently developed for the detection of HAV antigens and specific antibodies,162 culminating in the development of immunoglobulin M (IgM)-specific anti-HAV tests to distinguish recent HAV infection from a previous infection.223 In 1967, Deinhardt et al79 provided the first evidence for propagation of HAV in small primates, leading to the subsequent recovery of the CR326 strain of human HAV from an experimentally infected Saguinus mystax.252 In 1979, Provost and Hilleman304 were successful in cultivating and serially
passaging this HAV strain in cell culture—an important step that ultimately contributed to the development of a vaccine.
passaging this HAV strain in cell culture—an important step that ultimately contributed to the development of a vaccine.
Virus Classification
HAV is a nonenveloped, positive-sense, single-stranded RNA virus classified within the family Picornaviridae that otherwise include many medically and veterinary important pathogens subclassified into 12 genera based on genotypic and serologic characterization (Fig. 19.1). HAV differs from other picornaviruses by several attributes, as reviewed by Martin and Lemon249: (a) HAV is resistant to high temperatures, low pH, and drugs that inactivate many picornaviruses; (b) HAV replicates very slowly and generally without cytopathic effect in cell culture; (c) nucleotide and amino acid sequences, as well as predicted sizes of several HAV proteins are dissimilar; and (d) only one serotype of HAV has been identified, and one antigenic neutralization site is immunodominant. Therefore, HAV has been classified as the type species of a separate genus—Hepatovirus—and is still the only identified member of this genus. One other distinct species of picornavirus found in chickens and other birds, avian encephalomyelitis virus,251 was tentatively classified within the Hepatovirus genus on the basis of a close phylogenetic relationship to HAV.351 However, based on substantial molecular divergence in several proteins and its polyprotein translation mechanism, this virus has been assigned recently to a separate new genus within the Picornaviridae family—the Tremovirus genus.
Virus Host Range and Experimental Model Systems
HAV infects humans and nonhuman primates. Serologic and experimental infectivity studies in nonhuman primates using acute-phase sera from HAV-infected patients have revealed that HAV can be transmitted to chimpanzees (Pan troglodytes)78,82,256 and other Old World primates, such as vervet (Chlorocebus sp.), African green monkeys (Cercopithecus aethiops), rhesus and cynomolgus macaques (Macaca mulatta and M. fascicularis), as well as several species of New World primates, such as tamarins (Saguinus sp.), marmosets (Callithrix sp.), squirrel (Saimiri sp.), and owl monkeys (Aotus sp.).18,79,165,307 Disease in nonhuman primates resembles that in humans but is usually milder.307 There are several reports of isolation of HAV-related viruses from Old World monkeys in the wild, several of which have significant sequence variation and minor
antigenic differences with human HAV45,94,274,376 (see the Diversity of the Infectious Agent section). The simian virus isolated from an African green monkey caused severe hepatitis in African green monkeys, rhesus macaques, and tamarins but not in chimpanzees (although the virus replicated and the animals seroconverted).94 Correspondingly, after 20 passages in marmosets, human HAV MS-1 was more virulent for marmosets but was attenuated for chimpanzees.40 This suggests that host, species-specific variants may have been selected. To date, no rodent animal model has been shown to be susceptible to HAV, although experimental infection in guinea pigs resulted in virus replication in the absence of clinical disease.166 Chimpanzees, tamarins, and marmosets have been most extensively used as animal models of HAV infection.
antigenic differences with human HAV45,94,274,376 (see the Diversity of the Infectious Agent section). The simian virus isolated from an African green monkey caused severe hepatitis in African green monkeys, rhesus macaques, and tamarins but not in chimpanzees (although the virus replicated and the animals seroconverted).94 Correspondingly, after 20 passages in marmosets, human HAV MS-1 was more virulent for marmosets but was attenuated for chimpanzees.40 This suggests that host, species-specific variants may have been selected. To date, no rodent animal model has been shown to be susceptible to HAV, although experimental infection in guinea pigs resulted in virus replication in the absence of clinical disease.166 Chimpanzees, tamarins, and marmosets have been most extensively used as animal models of HAV infection.
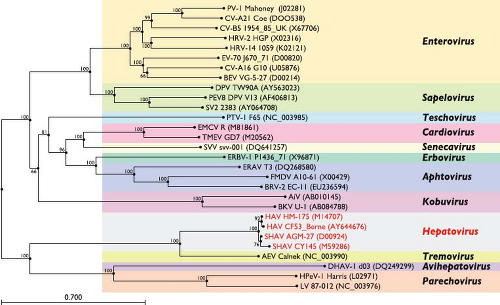 Figure 19.1. Phylogenetic relationship of hepatitis A virus (HAV) and simian HAV (SHAV) to representative species of other Picornaviridae genera. The relationship was examined by mid-pointed rooted neighbor-joining tree based on P1 (capsid) amino acid similarities using Clustal alignments (accession numbers for sequences of selected picornavirus strains are indicated in parentheses). Bootstrap values (%) are illustrated at nodes, and the genetic distance per amino acid per site is shown by the horizontal bar. The grouping in 12 genera is marked by colored boxes. HAV appears most closely related to tremoviruses (avian encephalomyelitis virus, AEV). |
HAV is usually difficult to adapt and grow in vitro. The virus was first isolated ex vivo in marmoset liver explant cultures and was subsequently propagated in continuous fetal rhesus monkey kidney cells304 and in a variety of different types of mammalian cells. Primary African green monkey kidney (AGMK) cells, primary human fibroblast cells, and continuous human diploid lung (MRC5) cells often have been used because of their suitability as potential vaccine substrates. Cell lines derived from AGMK (e.g., BS-C-1, Vero), fetal rhesus kidney (FRhK4), or human hepatoma (PLC/PRF/5) tissue also have been useful for studying virus replication and for propagating large amounts of virus.71,106,308 Surprisingly, HAV also replicates in cells of guinea pig, porcine, or dolphin origin.87
Wild-type HAV strains collected from infected patients usually replicate very slowly and to relatively low titers in cultured cells, often requiring weeks or even months and blind passages to reach maximal titers. With continued in vitro passage, however, the virus becomes progressively adapted to growth in cell culture, replicating more rapidly and achieving higher titers.57,59,71 During this process, the viral genome accumulates several mutations (see the Molecular Determinants of Hepatitis A Virus Adaptation to Cell Culture and Attenuation In Vivo section). Even when well adapted, HAV usually takes several days to weeks to reach maximal concentrations of propagated virus, achieving typical yields of 106 to 108 50% tissue culture infectious doses (TCID50)/mL of virus. Recently, a subpopulation of human hepatoma cells (Huh7) has been selected that support efficient growth of wild-type HAV strains without requiring further genetic adaptation.202 These cells may prove useful to characterize wild-type HAV strains isolated from patients and environmental samples.
In contrast to many other picornaviruses, HAV usually has few or no visible effects on the host cell. Thus, detection of HAV multiplication has usually required indirect methods: immunologic assays for viral antigens and hybridization or real-time polymerase chain reaction (RT-PCR) assays for HAV RNA. Radioimmunofocus or infrared fluorescent immunofocus assays can be used to quantify infectious HAV, select viral clones, and measure neutralizing anti-HAV antibodies.66,222
As with other positive-strand viruses, purified genomic RNA, whether extracted from virions or produced synthetically from cloned complementary DNA (cDNA) of cell culture-adapted HAV, is replication competent when transfected into permissive cultured cells.58 Accordingly, using synthetic RNA in cell culture and also applying findings from other picornaviruses, it has been possible to elucidate several features of the HAV replication strategy.
Morphology and Properties of Hepatitis A Virions
Virion Structure and Physicochemical Properties
HAV is a nonenveloped, approximately 27-nm spherical virus particle as first determined by Feinstone et al,101 who used immune-electron microscopy and negative staining techniques on stool specimens collected from acute-phase hepatitis A patients (Fig. 19.2). Mature HAV virions purified from feces collected from infected humans or chimpanzees band at 1.32 to 1.34 g/cm3 in cesium chloride (CsCl) and sediment at approximately 156S to 160S in neutral sucrose solutions.342 Additional populations of viral particles with different sedimentation characteristics have been isolated from human feces and cell culture. Particles with lower density that band at about 1.27 g/cm3 in CsCl and sediment at 70S to 80S are abundant in feces collected during early infection and probably represent empty capsids devoid of genomic RNA (also called procapsids) (see Fig. 19.2). Other premature or defective particles, including particles harboring capsid protein precursors, also can be observed.26,224,342 These alternative structures are
morphologically indistinguishable from typical HAV and have the same major surface antigens.
morphologically indistinguishable from typical HAV and have the same major surface antigens.
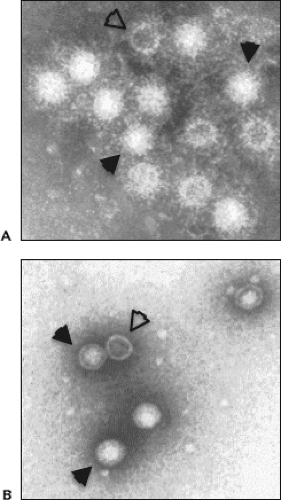 Figure 19.2. Hepatitis A virus (HAV) particles observed by electron microscopy. A: Immune electron micrograph of HAV particles from human stool heavily coated with and aggregated by human convalescent antibody. B: Negative staining showing 27- to 28-nm HAV particles with cubical symmetry purified from human stool. Both virions (closed arrowheads) and procapsids devoid of genomic material (open arrowheads) can be found. (Courtesy of Dr. Stephen M. Feinstone, Food and Drug Administration, Bethesda, MD.) |
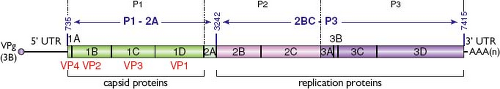 Figure 19.3. Hepatitis A virus (HAV) genome organization. The positive-strand RNA genome contains a single open reading frame (ORF), 5′ and 3′ untranslated regions (UTRs), is linked at its 5′ terminus to the virus-encoded protein 3B (also referred to as VPg), and is polyadenylated [AAA(n)] at its 3′ terminus. The ORF encodes capsid proteins 1A-D (also referred to as VP1-4 in the order indicated below the genome scheme; 1AB or VP4-VP2 precursor also is known as VP0), protein 2A that is involved in particle assembly as 1D-2A (VP1-2A) precursor, and proteins 2B-C and 3A-D involved in genome replication. Dotted lines delineate commonly termed P1, P2, and P3 coding segments in picornaviruses, whereas primary cleavage of HAV polyprotein generates P1-2A and 2BC-P3 polypeptides (which sequences are indicated by double-headed arrows). Numbering above the genome indicates nucleotide positions within the wild-type HM175 strain of HAV (GenBank accession number M14707). |
The HAV capsid encloses the viral genome, which is a linear, single-stranded, 33S RNA molecule of positive polarity approximately 7.5 kilobases (kb) in length.59,65,342 The viral genome encodes a single polyprotein that is subsequently cleaved primarily by the unique virus-encoded protease 3C to generate four capsid proteins (VP1-VP4) at its N-terminus and the nonstructural proteins involved in genome replication at its C-terminus (Fig. 19.3).
The HAV capsid contains 60 copies of the three major capsid proteins VP1, VP2, and VP3.120,342 Attempts to determine the atomic structure of HAV by X-ray crystallography have not been successful, although such studies have provided high-resolution images of virus particles from several other picornaviral genera. Medium-resolution images of the HAV particle, obtained by cryo-electron microscopy (Fig. 19.4) and generated by R. H. Cheng and collaborators (personal communication, 2011), suggest that there are prominent features at the icosahedral surface of the particle around fivefold and threefold axes of symmetry but no marked depression around the fivefold axes in contrast to the canyon found in enteroviruses.250
Hepatitis A Virus Resistance to Physical and Chemical Agents
In common with the enteroviruses, HAV is stable at low pH.327 It retains most of its infectivity when subjected to pH 1.0 for 2 hours at room temperature and is still infectious after 5 hours. The thermal stability of HAV, however, is considerably greater than that of other picornaviruses.344 Incubation of the virus for at least 4 weeks at room temperature (25°C) results in only a 100-fold decrease in infectivity.259,350 Significant loss of infectivity starts to occur with exposure at 60°C for short periods, and infectivity is destroyed almost instantaneously by heating above 85°C at neutral pH.285,305,344
HAV has been found to survive for days to months in experimentally contaminated fresh water, seawater, wastewater, soils, marine sediment, live oysters, and cream-filled cookies.350 Outbreaks of hepatitis A have been reported following ingestion of partially cooked bivalve mollusks, suggesting that even steaming may be insufficient to destroy the virus in this food source. In addition, HAV infectivity is highly resistant to drying, and infectious virus has been recovered from acetone-fixed cell sheets. The virus is also highly resistant to detergents, surviving a 1% concentration of sodium dodecyl sulfate, as well as to organic solvents such as diethyl ether, chloroform, or trichlorotrifluoroethane.294,344 Thus, solvent-detergent inactivation procedures do not reduce the infectivity of HAV, explaining why hepatitis A transmission has occasionally been associated with the administration of high purity, solvent-
detergent–treated clotting factor concentrates.227 These properties of the virus are likely to contribute significantly to its ability to spread through the environment and to cause common-source outbreaks of hepatitis.
detergent–treated clotting factor concentrates.227 These properties of the virus are likely to contribute significantly to its ability to spread through the environment and to cause common-source outbreaks of hepatitis.
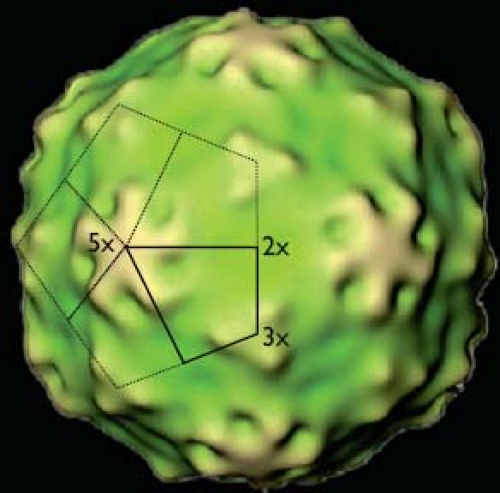 Figure 19.4. Medium-resolution image of the structure of the hepatitis A virus (HAV) particle as revealed by cryo-electron microscopy. The solid kite defines a protomer that is likely occupied by single copies of each capsid protein VP1, VP2, and VP3. Five copies of such a protomer are linked at the fivefold (5×) axis of symmetry to form a pentamer assembly subunit. The mature particle contains 12 pentamers arranged on an icosahedral surface or 60 copies of each individual capsid protein. Threefold (3×) and twofold (2×) axes also are indicated. Coloring is based on the distance from the center of the particle, with yellow depicting exposed regions at the surface of the particle. (Courtesy of Profs. R. Holland Cheng, University of California at Davis, Davis, CA, and Tatsuo Miyamura, National Institute of Infectious Diseases, Tokyo, Japan.) |
Inactivation of HAV was reviewed by Coulepis et al.63 Appropriate measures rely on autoclaving (121°C for 20 minutes), exposure to chlorine-containing compounds (sodium hypochlorite, 3–10 mg/L at 20°C for 5–15 minutes or free residual chlorine concentrations of 2.0–2.5 mg/L for 15 minutes),294 or to a quaternary ammonium formulation containing 23% hydrogen chloride (HCl). HAV also is inactivated by incubation in 3% formaldehyde for 5 minutes, or in 70% ethanol for 60 minutes, and by β-propiolactone or ultraviolet irradiation.258
Genome Structure
Genomic Organization
Similar to all picornaviral genomes, the HAV genome is a single-stranded RNA molecule of positive polarity that can be divided into three parts (see Fig. 19.3): (a) a relatively lengthy 5′ untranslated region (UTR) that does not have a cap structure at its 5′ end but instead has a 2.5 kDa, covalently bound, virus-encoded protein—VPg (also known as 3B)389; (b) a single, large open reading frame (ORF) encoding a polyprotein of approximately 2,227 amino acids in length that is proteolytically processed into structural (P1-2A) and nonstructural (2B-C and P3 or 3A-D) viral polypeptides; and (c) a short 3′ UTR of 63 nucleotides that is followed by a poly(A) tail of varying lengths (40–80 nucleotides) typical of picornavirus genomes. Complete sequence lengths range from 7,470 to 7,487 nucleotides excluding the 3′ poly(A) tail.
The HAV genome is very rich in A+U bases (62%) as compared to the median A+U base composition of 54% found among other picornaviruses—a feature that is likely to affect base-pair composition and biologically relevant folds of the genome. As in other picornaviruses, however, the HAV genome also is predicted to exist as a circular entity with both 5′ and 3′ ends in close proximity to facilitate RNA translation to replication switches.284
Untranslated Regions
The nucleotide sequence of the HAV genome begins at the 5′ end with two uridyl residues typical of picornaviruses. The 5′ UTR is approximately 729 to 749 nucleotides long and has a high level of secondary RNA structure comprising several complex stem-loop structures (Fig. 19.5). RNA secondary structure and tertiary interactions (pseudoknots) within this segment of the genome have been established by a combination of (a) phylogenetic comparative sequence analyses to predict thermodynamically stable base pairing, (b) functional genetic studies, and (c) direct biophysical and nuclease mapping techniques with single- or double-strand–specific ribonucleases (RNases).41,215,336 Among HAV strains, the 5′ UTR appears to be the most conserved region of the genome (≥89% nucleotide identity among strains representing genotypes I, II, and III).41 All picornavirus genomes have secondary structural elements at their extreme 5′ end that differ in length and folding.284 In the case of HAV, the 5′ UTR has an unbranched terminal stem of very low free energy comprising the 41 5′ terminal nucleotides. This element (stem-loop I; see Fig. 19.5) is likely to be important for RNA replication.
As in all picornaviruses, the most dominant structural unit in the HAV 5′ UTR is the internal ribosome entry site (IRES), which determines the initiation of viral translation in a 5′ cap-independent fashion (see Fig. 19.5). Depending on structural similarities at the secondary and tertiary levels, and the nature of cellular translation factors required to initiate translation, picornaviral IRESs are divided into four groups.284 The HAV IRES makes up a group on its own, designated type III IRES, which is distinct from all other known IRESs.20,284 The boundaries of the HAV IRES were mapped by using bicistronic messenger RNAs (mRNAs) comprised of a first reporter gene followed by various segments of the HAV 5′ UTR that controlled translation of a second, downstream reporter gene. Characterization of their translation activities in reticulocyte lysates or in cell culture revealed that the IRES is located between nucleotides 154 and 735.43,125
Between the 5′ terminal stem-loop I and the IRES lies a sequence of approximately 110 nucleotides (nucleotides 42–154) that includes (a) two pseudoknots (IIa and IIb, nucleotides 42–94),336 the sequence of which is highly conserved among HAV strains, and (b) a partially ordered polypyrimidine tract and a single-stranded region (nucleotides 95–154),149 which are susceptible to naturally occurring mutations, particularly deletions.41 This 5′ spacer domain corresponds to the most variable segment in length and sequence among all picornaviruses.284 These sequences may function as replication elements and/or contribute essential virus–host interactions.
The 3′ UTR of HAV RNA shows a high proportion of differences between HAV strains (up to 20%). It has been less extensively studied than the 5′ UTR but also has a high propensity to form higher-order structures. Hypothetical structural models of the HAV 3′ UTR have been proposed based on probing with specific RNases and computer-aided predictions, suggesting that a pseudoknot structure is favored85 (e-Fig. 19.1).
Structural Proteins and Replication Proteins
Uniquely among Picornaviridae genera, the primary cleavage of the HAV polyprotein occurs between the P1-2A and 2BC-P3 segments of the polyprotein under the direction of the only protease encoded by the virus—the 3C protein184,248 (see Fig. 19.3). The P1-2A segment comprises four structural polypeptides (in order): VP4 (1A), VP2 (1B), VP3 (1C), and VP1 (1D), named according to their homologs in the poliovirus capsid based on relative molecular masses, with VP1 being the largest. These proteins are approximately 23, 222, 246, and 274 amino acids in length, respectively (Table 19.1).59,138,247 VP1, however, has a heterogeneous carboxy terminus,138 reflecting a unique maturation mechanism. Whereas VP2, VP3, and VP1 comprise the viral capsid, VP4 is substantially smaller than its homologs in other picornaviruses and has never been experimentally determined to be part of the HAV capsid. The 2A protein of HAV does not encode a primary cleavage function as found in the 2A proteins of most other picornaviruses, neither is it involved in genome replication.250 HAV 2A functions in capsid assembly as a fusion precursor with VP160 and remains attached to some otherwise fully formed virions.9 In-frame insertions within the HAV genome of exogenous protein coding sequences, flanked by 3C cleavage sites at the 2A/2B junction, has resulted in replication-competent virus, further
confirming that 2A has no function in cis with the remainder of the nonstructural proteins.21
confirming that 2A has no function in cis with the remainder of the nonstructural proteins.21
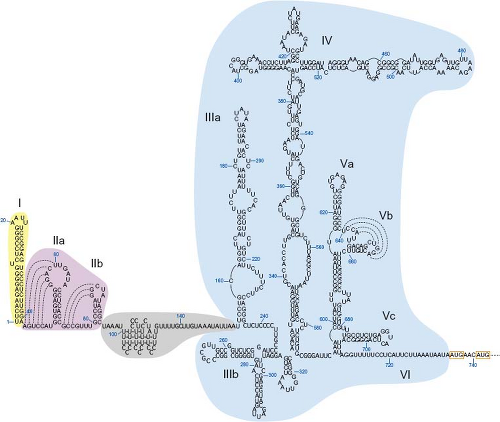 Figure 19.5. Proposed RNA structure of the 5′ untranslated region (5′ UTR) of hepatitis A virus (HAV; wild-type HM175 strain, accession number M14707). Nucleotides (in blue) are numbered according to their positions within the HAV genome. Subdomains comprised of stem-loop structures are numbered sequentially (Roman numerals). Putative tertiary pseudoknots are represented by dotted lines to indicate possible interactions between nucleotides. The 5′ terminal stem-loop structure (I), pseudoknot elements (IIa-b), and polypyrimidine tract/single-stranded region (see text) are represented by yellow, pink, and gray–shaded areas, respectively. The internal ribosome entry site (IRES) is delineated by the blue-shaded area and comprises RNA structures IIIa to VI (nucleotides 154–735). The two in-frame AUG initiator codons of the viral polyprotein are boxed. (Modified from Martin A, Lemon S. The molecular biology of hepatitis A virus. In: Ou J-H, ed. Hepatitis Viruses. Norwell, MA: Kluwer Academic Publishers; 2002:23–50, with kind permission from Springer Science+Business Media B.V.) |
Nonstructural proteins derived from the 2BC-P3 segment of the polyprotein probably all contribute directly to the assembly of the membrane-bound viral replicase complex (see Table 19.1). Proteins 2B and 2C, and probably also the precursor 2BC, are involved in directing the rearrangements of cellular membranes required for replicase assembly.130,369 It has been suggested that 2B, containing hydrophobic sequences, anchors the replicase complexes to altered intracellular membranes.130 2C has NTPase activity126 and contains an RNA helicase sequence motif.84
Among the P3 nonstructural proteins, 3D is assumed to be the viral RNA-dependent RNA polymerase, although no direct data exist. 3C has been crystallized and is a chymotrypsin-like cysteine protease3,23,409 (Fig. 19.6). It appears to be the only virus-encoded protease of HAV, which is responsible for all co- or posttranslational cleavage events within the HAV polyprotein except for those at VP4/VP2 and VP1/2A junctions.151,242 In addition, HAV 3C protease and/or its precursors 3ABC and 3CD also appear to cleave cellular IRES-transacting factors and adaptor molecules acting in interferon signaling pathways (see the Infectious Viral Life Cycle and the Pathogenesis and
Host Immune Responses sections). P3 also generates protein 3B (also referred to as VPg), which is attached to the 5′ end of both positive- and negative-strand RNAs,389 and protein 3A, which comprises a hydrophobic 21 amino acid stretch that is believed to anchor the 3AB precursor of VPg (3B) to cellular membranes and to target the 3ABC precursor to mitochondrial membranes.404
Host Immune Responses sections). P3 also generates protein 3B (also referred to as VPg), which is attached to the 5′ end of both positive- and negative-strand RNAs,389 and protein 3A, which comprises a hydrophobic 21 amino acid stretch that is believed to anchor the 3AB precursor of VPg (3B) to cellular membranes and to target the 3ABC precursor to mitochondrial membranes.404
Table 19.1 Hepatitis A Virus Structural and Nonstructural Proteins | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Diversity of the Infectious Agent
Genetic Diversity
HAV identification and initial characterization were done with strain MS-1 isolated in 1964 from the blood and stools of a patient in New York.101,204 In the mid-1980s, the first comparative study of the nucleotide sequences of HAV performed by RNase T1 oligonucleotide mapping of eight strains originating from diverse geographic sources revealed a variation in the order of magnitude reported for other picornaviruses.390 Several other laboratories reported nucleotide sequencing of full-length genomes from several HAV strains isolated directly from the stools of patients (e.g., HM175; Australia, 1976),59,71 or after a few passages in small primates or cell culture (e.g., LA; USA, 1976),275 MBB (North Africa, 1978),286 and GBM (Germany, 1976).136 Wild-type strain HM175 has been adapted to yield the widest range of mutants in cell culture, including attenuated, persistently infecting, cytopathic and neutralization-resistant variants (see the Molecular Determinants of Hepatitis A Virus Adaptation to Cell Culture section). Attenuated mutants of HM175,193 CR326 (Costa Rica, 1960),231 H2 (China, 1982),244 and variants of strains GBM384 and MBB167 are the best characterized candidates for live attenuated vaccines,
as reviewed by Flehmig et al,107 Hu et al,168 and Provost et al303 (see the Hepatitis A Vaccines section).
as reviewed by Flehmig et al,107 Hu et al,168 and Provost et al303 (see the Hepatitis A Vaccines section).
 Figure 19.6. Ribbon representation of the crystal structure of hepatitis A virus (HAV) 3C protease (PDB accession 2HAL).409 A: Ribbon is colored by secondary structure with red, helix; yellow, sheet; green, loop. Side chains of the catalytic triad (D84, H44, C172) are shown as spheres across the top of the active site. The side chain of H191, the primary residue responsible for the glutamine specificity in P1, is also shown as spheres. B: A stick representation of the LFFE-FMK inhibitor, which mimics substrate residues P1-P4, is shown bound in the active site. Spheres and sticks are colored by atom type. (Courtesy of Dr. Bruce A. Malcolm, Johnson and Johnson Infectious Diseases and Vaccines, Beerse, Belgium.) |
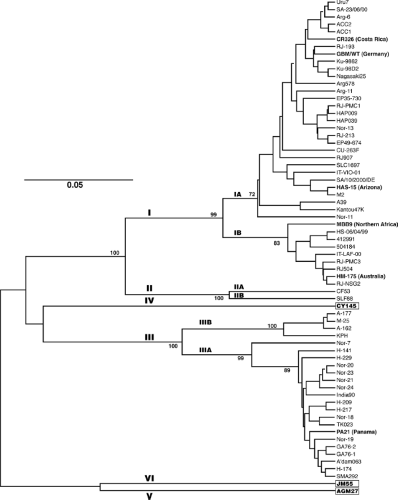 Figure 19.7. Phylogenetic tree based on hepatitis A virus (HAV) sequences (168 nucleotides) in the 2A region. Statistically significant bootstrap values (%) are given at the nodes, and genetic distance per nucleotide per site is shown by the horizontal bar. Genotypes are indicated by Roman numerals, whereas subgenotypes are indicated by A and B. The country of origin is indicated for best characterized strains of genotypes I, II, and III. The prototype MS-1 strain of HAV (not shown) belongs to subgenotype IA. The only known representatives of genotypes IV, V, and VI were collected from Old World monkeys. (Modified from Lu L, Ching KZ, de Paula VS, et al. Characterization of the complete genomic sequence of genotype II hepatitis A virus (CF53/Berne isolate). J Gen Virol 2004;85:2943–2952, with permission.) |
The genetic diversity of HAV has further been investigated by determining the partial genomic nucleotide sequences of 152 HAV strains recovered from various human or simian sources and geographical areas234 (Fig. 19.7). Following virus purification by capture with an HAV monoclonal antibody, reverse transcription of viral RNA, and amplification by RT-PCR,181 genotypic analysis was based on the nucleotide sequence of a short genomic segment (168 nucleotides) that encompassed the relatively variable 2A coding sequence (originally thought to span the VP1/2A junction).
Additional phylogenetic studies were more recently carried out using a wider range of HAV isolates that were underrepresented in the initial genotyping studies. This included HAV strains from South America, North and Central Africa, and India. In addition, full-length VP1 sequences (900 nucleotides) were chosen to establish nucleotide comparisons, considering that several residues of VP1 contribute to the major immunodominant site of HAV. Overall strain variation in the complete VP1 gene was found to be higher than 20% at the nucleotide level and around 10% at the amino acid level.62
The current view is that HAV circulates worldwide as a total of six genotypes (≤85% identity) with three genotypes (I, II and III) subdivided into subtypes A and B (86%–92.5% identity within subtypes)234 (see Fig. 19.7). Genotypes I, II, and III comprise all human strains, among which subgenotype IA and genotype III are the most prevalent worldwide (e-Fig. 19.2). Each of the three simian HAV genotypes are defined by a single strain isolated from naturally infected Old World monkeys, cynomolgus macaques (genotypes IV and VI),274,318 and African green monkeys (genotype V).376 All simian HAV strains have a typical signature sequence at the VP3/VP1 junction that distinguishes them from human strains.42,318,376
Clusters within genotypes predominate in certain geographic regions (see e-Fig. 19.2), such as a group of subgenotype
IA strains with more than 97% identity that represent nearly all of the viruses from the United States, suggesting endemic spread. Other regions, such as Europe and Japan, have a greater genetic mixture of viruses, suggesting regular introduction of strains by travelers returning from endemic regions. A higher degree of heterogeneity than reported previously has been found in strains isolated in South America.67
IA strains with more than 97% identity that represent nearly all of the viruses from the United States, suggesting endemic spread. Other regions, such as Europe and Japan, have a greater genetic mixture of viruses, suggesting regular introduction of strains by travelers returning from endemic regions. A higher degree of heterogeneity than reported previously has been found in strains isolated in South America.67
To better define the HAV mode of evolution in populations, full-length VP1 sequences of a collection of genotype IA HAV strains isolated in France from 1984 to 2001 were analyzed. This revealed a mean rate of 9.76 × 10−4 nucleotide substitutions per site per year, and an estimation of a synonymous substitution rate around 5 × 10−3 substitutions per site per year. The latter is clearly lower than that observed for other representative members of the family Picornaviridae and may be related to the slower replication cycle of HAV, its particular tropism, or its transmission mode.265
Virion Antigenic Structure and Diversity
Comparisons of the nucleotide sequences of various human HAV strains from widely separated geographic regions have demonstrated high amino acid conservation in the sequences of the viral capsid proteins (>70%). Likewise, viruses belonging to distinct genotypes elicit antibodies with substantial cross-neutralizing activity, suggesting that there is only one HAV serotype among human strains.353 A primate HAV strain recovered from a captive New World monkey (owl monkey) was considered to be a human (genotype IIIA) rather than a simian strain following biological characterization and sequence analysis42,318 (see e-Fig. 19.2). Conversely, in strains of HAV isolated from naturally infected Old World monkeys (cynomolgus and African green monkeys), some monoclonal antibodies are capable of distinguishing unique epitopes, indicating some degree of antigenic singularity in simian strains.274,376 However, whereas African green monkey virus from genotype V caused no disease in chimpanzees, it elicited an anti-HAV response that protected the chimpanzees against disease after challenge with the virulent human-derived HM175 strain.94 These observations suggest that even simian and human strains of HAV demonstrate substantial antigenic cross-reactivity.
Neutralization epitopes of HAV are conformational and generally are not displayed on isolated HAV proteins. Correspondingly, neutralizing murine monoclonal antibodies do not recognize denatured capsid proteins, and antisera raised against proteins expressed by recombinant DNA techniques show only weak reactivity with native capsids and have very limited virus neutralization activity.170 Most murine monoclonal antibodies to HAV compete with each other and are able to substantially block the binding of polyclonal human antibodies to virus in solid-phase competition immunoassays.171,354 Similarly, human and chimpanzee neutralizing monoclonal anti-HAV antibodies recovered by phage display have been shown to compete with murine monoclonal antibodies.199,326 This suggests that there may be only a limited number of antigenic epitopes that are closely clustered on the surface of the virus.
Antigenic variants of HAV that resist neutralization by murine monoclonal antibodies have been selected in cell culture by continued passage of HM175 virus in the presence of these antibodies.354 Sequencing of the genomic regions encoding their capsid proteins revealed single amino acid changes in each mutant with a limited number of changes confined to VP3 (two mutations) and VP1 (four mutations).296,297 Similarly, in strain HAS-15 (Arizona, 1979), a small number of mutations that confer resistance to neutralizing antibodies also were selected in VP3 and VP1.273 Competition studies with these mutants confirmed the existence of an immunodominant site composed of residues of VP1 (at positions 102, 171, and 176) and VP3 (at positions 70 and 74) that are likely to be closely positioned on the surface of the HAV particle and reside on exposed loops that connect β strands of the capsid proteins. This feature is likely to distinguish HAV from enteroviruses, for which equivalent residues do not lie in close vicinity at the surface of the capsid.160 A crystallographic structure of the HAV capsid would shed light on the conformational nature of the immunodominant antigenic site. An apparently distinct site involving C-terminal residues of VP1 has been identified from HM175 neutralization escape mutants.297 In addition, two continuous epitopes near the N-terminus of VP1 (residues 11–25) and within VP3 (residues 110–121), which elicit weak neutralizing activity as peptides also may represent secondary neutralization sites.34,95 In the case of poliovirus, the VP1 N-terminus is internally located in the virion capsid but is exposed during virus attachment to cells.44
The finding of limited amino acid changes among multiple neutralization escape mutants suggests that there are important constraints to the antigenic or structural variations of HAV. Recently, however, several HAV variants bearing substitutions or deletions at or around the immunodominant neutralization site or secondary neutralization sites were isolated in sporadic cases or during outbreaks of hepatitis A. These strains showed at least partial resistance to antiserum generated against HAV vaccine or to murine monoclonal anti-HAV antibodies, suggesting a potential for the emergence of HAV antigenic variants or new serotypes in the population.62,119,123,292,323
Infectious Viral Life Cycle
Attachment and Entry
HAV has been shown to bind to a wide range of cultured animal cells, including nonpermissive cells. Although nothing is known about viral determinants involved in HAV cell attachment, it has been observed that mature virions attach relatively rapidly and in a calcium-dependent way25,352,394,412 but undergo comparatively slow uncoating with release of viral RNA into the cytoplasm about 4 hours postinfection.27
A candidate cellular receptor (HAVcr1), a mucin-like class 1 integral membrane glycoprotein of 451 amino acids370 (Fig. 19.8A), was first identified in HAV-susceptible AGMK cells using an expression cloning strategy and a monoclonal antibody that blocked binding of HAV to the cell surface.191 A molecule with 95% amino acid similarity was concomitantly identified in an HAV-susceptible hybrid marmoset-Vero cell line.14 A human homolog of HAVcr1 (with 79% identity) was later shown to interact with cell culture–adapted HAV.100 The N-terminal cysteine-rich immunoglobulin-like region of the HAVcr1 ectodomain is sufficient for binding HAV, but both this region and the mucin-like region are required for viral particle conformational changes leading to HAV uncoating346 (Fig. 19.8A,B). A soluble form of HAVcr1 also is able to neutralize wild-type HAV,345 providing additional evidence for the role of this molecule as the HAV receptor in vivo. Recent
studies have identified TIM-1, an atopy susceptibility gene, as the gene that encodes HAVcr1.261 The T-cell immunoglobulin mucin (TIM) gene family, particularly TIM-1, are cell surface receptors that are important in T-cell regulation and Th-cell differentiation (see the Clinical Features section). HAVcr1 is widely distributed in different tissues100; thus, the tropism of HAV for the liver remains an enigma waiting to be resolved.
studies have identified TIM-1, an atopy susceptibility gene, as the gene that encodes HAVcr1.261 The T-cell immunoglobulin mucin (TIM) gene family, particularly TIM-1, are cell surface receptors that are important in T-cell regulation and Th-cell differentiation (see the Clinical Features section). HAVcr1 is widely distributed in different tissues100; thus, the tropism of HAV for the liver remains an enigma waiting to be resolved.
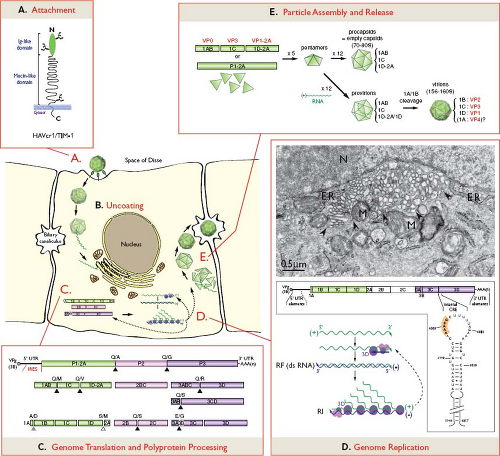 Figure 19.8. Hepatitis A virus (HAV) infectious life cycle. A: HAV enters hepatocytes by attachment to a cellular receptor, likely HAVcr1/TIM-1. This receptor, as schematically represented, comprises an N-terminal immunoglobulin-like domain and a mucin-like domain with three N-glycosylated sites (branched elements).370 B: Details of HAV uncoating remain largely unknown. C: Once released within the cell cytoplasm, virion RNA serves as a template for translation of the viral polyprotein, which is subsequently cleaved as depicted. Cleavages are carried out by HAV protease 3C at most junctions (black triangles), likely by a cellular protease at the 1D/2A junction (open triangle), or upon RNA encapsidation at the 1A/1B junction (gray triangle). Dipeptides representing cleavage sites between stable polypeptide precursors or mature proteins are indicated above corresponding boxes. D: Genome replication subsequently takes place within the virus-induced tubular vesicular network. The electron micrograph of FRhK-4 cells infected with cell culture–adapted HAV demonstrates rearrangement of cytosolic membranes into a network of tubular-vesicular structures (delineated by arrowheads), which appear closely associated with membranes of the endoplasmic reticulum (ER) and mitochondria (M) in the vicinity of the nucleus (N).130 The positive-strand RNA is used as a template by the viral replicase complex comprised of polymerase 3D and other nonstructural proteins for minus-strand RNA synthesis, thereby generating replicating forms (RF). The minus-strand RNA serves in turn as a template for the synthesis of multiple positive-strand RNAs (replication intermediates, RI). Cis-active RNA elements (CRE) located at the 5′ and 3′ terminal regions of the genome, as well as within the 3D coding sequence are required for RNA replication. The stem-loop structure of the internal CRE, as depicted below the genome, includes a relatively lengthy stem (nucleotide numbering from HAV full-length genome) and a large loop that contains a motif boxed in orange, typical of all picornaviral CREs and necessary for their function.405 Newly synthesized positive-strand RNAs can be either engaged in translation, replication, or encapsidation, as represented by dashed arrows within the cell. (Electron micrograph courtesy of Dr. Rainer Gosert, Institute for Medical Microbiology, Basel, Switzerland.) E: Virion assembly is likely to proceed through indicated assembly intermediates, ultimately leading to the production of infectious virions, which are subsequently released into biliary canaliculi. |
A surrogate entry mechanism of HAV has been proposed via immunocomplexes with HAV-specific immunoglobulin A (IgA), which are present in significant amounts during acute infection. This was based on the observation that HAV/IgA complexes can be endocytosed in hepatocyte cultures via the hepatocellular asialoglycoprotein receptor (ASGPR), which mediates uptake of IgA.88 IgA-mediated transcytosis of HAV via polymeric immunoglobulin receptor also was observed in vitro and proposed as a mechanism for HAV to overcome the epithelial barrier of the intestinal tract.86
Genome Translation, Polyprotein Processing, and Replication
The entire HAV life cycle occurs in the cytoplasm of the cell (Fig. 19.8). HAV genome translation and replication appear to take place in association with a tubular-vesicular membranous network specifically rearranged from cytosolic membranes by proteins 2B, 2C, or 2BC in HAV-infected cells,130,369 as is the case for other picornaviruses (Fig. 19.8D). The HAV 2B protein is localized predominantly in the endoplasmic reticulum (ER),76 suggesting that the tubular vesicular structures may be derived from the ER.197 Unlike enterovirus homologous proteins, HAV 2B has little, if any, effect on ER and Golgi calcium homeostasis, or on protein trafficking through the secretory pathway,76 which is consistent with the hypothesis that hepatitis A virions may use the secretory pathway to exit the cell.28
Translation
Uncoated RNA is translated via an IRES-dependent mechanism that involves cellular factors so that 40S ribosomal subunits bind and begin translation at the correct initiation codon and not at multiple AUG codons scattered throughout the 5′ UTR. This mechanism contrasts with typical eukaryotic translation, in which 40S subunits recognize an m7G-cap at the 5′ mRNA terminus (not present in the HAV genome) and then scan for the nearest AUG within an appropriate sequence motif.203 Initiation of HAV translation may start at either of two in-frame AUG codons at positions 735 through 737 and 741 through 743366 (see Fig. 19.5). Although either codon could function independently, selective deletion experiments have demonstrated that the second of these codons is preferentially used in vitro and in cells.366
The HAV IRES is far less efficient for directing translation in acellular systems and in various cell lines than are encephalomyocarditis virus (EMCV) and foot-and-mouth disease virus (FMDV) IRES, for example.32,43,125,396 Inefficient translation is likely owing to the fact that the HAV IRES uniquely requires intact cellular initiation factor eIF-4G to function, unlike other picornavirus IRESs that utilize a cleaved fragment of this factor,31 and that it probably competes poorly for this and other necessary cellular factors. HAV translation and replication are thus likely to depend on continued synthesis of host factors. Consistent with this hypothesis is the fact that HAV generally does not shut off host protein synthesis, unlike other picornaviruses. In the context of virus/cell competition, HAV appears to have adopted a naturally biased, deoptimized codon usage with respect to that of its cellular host.12,322 It has been hypothesized that this finely tuned strategy, observed specifically in the capsid coding region, might avoid competition between HAV and the host cell for cellular transfer RNAs (tRNAs), modulate translation kinetics, and allow proper protein folding to reach the highest viral fitness.12
HAV IRES activity appears to be modulated by several cellular proteins, including polypyrimidine tract-binding protein (PTB),51,129 glyceraldehyde 3-phosphate dehydrogenase (GAPDH),51,331,408 poly(rC) binding protein 2 (PCBP2),133 and lupus autoantigen (La).61 GAPDH and PTB compete for binding to overlapping sites in the IRES (stem-loop III at the 5′ end of the IRES)331 (see Fig. 19.5). Although the mechanisms by which these cellular proteins modulate the translational activities of HAV IRES are unknown, it has been speculated that GAPDH may have a destabilization effect on RNA secondary structure, leading to translation suppression,408 whereas PTB may have a stabilizing action on higher-ordered RNA structures, thereby reversing GAPDH action and stimulating translation.129,408 In support of this hypothesis, a naturally occurring mutation that enhances translation in AGMK cells332 decreases the affinity of GAPDH for stem-loop III in the IRES.408
Polyprotein Processing
Hepatoviruses follow a unique cleavage cascade distinct from all other picornavirus genera, as reviewed in Martin and Lemon.249 The primary, co-translational cleavage during HAV polyprotein translation is mediated by the unique virus-encoded protease (3C) at the 2A/2B junction, thus releasing a P1-2A precursor of 836 amino acids from the downstream nonstructural proteins128,248 (Fig. 19.8C). In most picornavirus genera, the primary polyprotein cleavage also occurs at the 2A/2B junction, although by a different mechanism; a unique sequence of amino acids (Asn-Pro-Gly-Pro) located at the 2A/2B junction possesses an inherent propensity to induce ribosome skipping, resulting in “cleavage” of the nascent polyprotein between Gly and Pro residues. Enteroviruses encode a second protease (2A) that autocatalytically cleaves its own N-terminus at the P1/2A junction.284
The key protease 3C carries out most other cleavages in the HAV polyprotein,151,182,242,330,368 as in other picornaviruses. The 3C-specific cleavage sites are formed by at least six residues surrounding the scissile bonds (amino acids at positions P4-P1 upstream, and P′1-P′2 downstream, of the cleavage site). HAV 3C is in general less discriminating than other picornaviral 3C proteases and recognizes Gln or Glu residues at position P1, any small residue or even charged residues with large side chains at position P′1 (e.g., Gln/Arg at the 3C/3D site), virtually any amino acid at position P′2, and residues with large side chains at position P4241 (see Figs. 19.6 and 19.8C). The 3C-mediated cleavages occur according to preferential site order and also release stable intermediate precursors, such as 2BC, 3ABC, 3CD that appear to contribute specific functions in the HAV life cycle (see Table 19.1).210,301,310,404
Cleavage at two junctions, VP1(1D)/2A and VP4(1A)/VP2(1B), is not carried out by the 3C protease. Uniquely among picornaviruses, VP1 is apparently released from 2A by cellular proteases.138,247 The C-terminus of VP1 is heterogeneous,138 with a predominant form releasing a 2A protein of
71 amino acids. It has been proposed that, as for Mengovirus, cellular proteases may trim 2A sequences from VP1-2A to generate VP1 intermediate forms, as well as mature VP1 protein.138,247 The maturation cleavage at the VP4 (1A)/VP2 (1B) junction, similar to other picornaviruses, takes place in late stages of virion morphogenesis (see Fig. 19.8E) and occurs by an unknown, perhaps autocatalytic, mechanism to yield capsid proteins VP4 and VP2.
71 amino acids. It has been proposed that, as for Mengovirus, cellular proteases may trim 2A sequences from VP1-2A to generate VP1 intermediate forms, as well as mature VP1 protein.138,247 The maturation cleavage at the VP4 (1A)/VP2 (1B) junction, similar to other picornaviruses, takes place in late stages of virion morphogenesis (see Fig. 19.8E) and occurs by an unknown, perhaps autocatalytic, mechanism to yield capsid proteins VP4 and VP2.
RNA Replication
Within the membrane-bound replicase complex, the virion RNA serves as a template for negative-strand RNA synthesis by the RNA-dependent, RNA polymerase 3D, generating double-stranded replicative forms (dsRNA) (Fig. 19.8D). This negative-strand intermediate then acts in turn as a template for the synthesis of multiple positive-strand RNA molecules, yielding replication intermediates (RIs). This asymmetric process suggests that mechanisms of replication of positive and negative strands may show some differences. Newly synthesized positive-strand RNAs can be translated into proteins, replicated, or packaged into new virions.
There are several genomic cis-active RNA elements (CREs) required for the initiation of viral RNA replication. Terminal sequences and/or structures present at the 5′ and 3′ ends of the genome, as well as the poly(A) 3′ terminal tail are likely to contain important replication elements. Consistent with this hypothesis, HAV genome-length RNAs that lacked the two 5′ terminal nucleotides (UU) or nucleotides 2 and 3 (UC), which are thought to be involved in a stem-loop structure (see Fig. 19.5), were not infectious.150 One study suggested that elements required for RNA replication may extend downstream of nucleotide 151 of the 5′ UTR (i.e., beyond the polypyrimidine tract), because a genome containing only the 5′ terminal 151 nucleotides of the 5′ UTR followed by the EMCV IRES was noninfectious, in contrast to chimeric genomes containing the first 237 HAV nucleotides fused to EMCV IRES.185 Likewise, whereas deletion of the pyrimidine-rich tract (nucleotides 96–139) did not alter viral replication in cell culture, deletions that also included nucleotides 140 through 144 immediately downstream of it (in a predicted single-stranded region (see Fig. 19.5) yielded a temperature-sensitive virus336 with defective RNA synthesis.338 The importance of the poly(A) 3′ terminal tail has been corroborated by observations that viral replication is delayed in cells transfected with genomes lacking part or all of the poly(A) sequence, and the poly(A) tail is concomitantly regenerated in progeny virus.212
In addition, other internal CREs first identified in a rhinovirus genome262 have been subsequently identified in several other picornavirus genera and are often located within genomic coding sequences. These elements comprise stem-loop structures of variable lengths with a common apical loop containing an “AAAC” motif.356 In poliovirus, the CRE was shown to template the uridylylation of 3B (VPg)—the small genome-linked protein—generating VPg-pUpU that functions as a protein primer for viral RNA synthesis.287 It is still debated whether both negative- and positive-strand RNA synthesis require CRE-dependent VPg uridylylation or whether negative-strand synthesis can be primed by VPg.356 For HAV, a CRE has been identified in the 3D coding sequence by combined phylogenetic and thermodynamic predictive strategies, as well as mutational analyses.405 HAV CRE comprises a larger stem loop with lower free-folding energy than CREs of other picornaviruses (see Fig. 19.8D), suggesting that there are likely to be important differences in the details of the VPg uridylylation templated process for HAV. This is further supported by the apparent targeting of HAV 3A (and likely also the 3AB precursor of VPg) to mitochondrial rather than ER membranes.389,404 Evidence suggests that internal CREs function coordinately with the other cis-active RNA sequences and structures at the 5′ and 3′ termini of picornavirus RNAs to mediate RNA replication, presumably via ribonucleoprotein interactions that might be facilitated by the NTPase activity of 2C.356 Along these lines, interactions between HAV proteins 3C and 3AB with structures formed at both termini of the HAV genome have been documented in vitro.22,211,213,293
Translation to Replication Switches
Interestingly, several cellular proteins may be involved in the finely tuned regulation of HAV RNA translation to replication switches, given that the same genomic molecule may be engaged in either process and that viral polysomes must be cleared of ribosomes before RNA replication can occur. Several sites for binding of GAPDH in the HAV 3′ UTR have been described.85 Therefore, binding of GAPDH to both 5′51,331,408 (see earlier discussion) and 3′ termini of the HAV genome might promote the establishment of their close spatial proximity and have a role in RNA translation-replication switches. Viral protein and RNA syntheses also might be coordinated by the binding of full-length PCBP2 to the polypyrimidine tract of the 5′ UTR, whereas a truncated form of this cellular protein, which is cleaved by HAV 3C protease, has lesser affinity for HAV RNA, thus possibly facilitating the switch toward replication.414 Similarly, another IRES trans-acting factor—PTB129—is cleaved upon overexpression of HAV 3C protease, resulting in translation inhibition and hypothetically replication turn on.189 However, the biological relevance of these observations is uncertain, because cleavage of these cellular factors has not been confirmed in HAV-infected cells, maybe as the result of low 3C quantities being present owing to protracted virus growth. HAV IRES was shown to be stimulated by polyadenylation in an acellular system, in contrast to FMDV IRES, suggesting a possible role for the 3′ terminal poly(A) residues of the genome in regulating HAV translation.290 More recently, poly(A)-binding protein (PABP) was shown to interact with polyadenylated HAV 3′ UTR.413 This could hypothetically allow the bridging of HAV IRES via PABP-eIF4G interaction, resulting in the circularization of the RNA, such as in poliovirus and cellular mRNAs.158 PABP also is targeted by HAV 3C protease in HAV-infected cells yielding a C-terminally truncated polypeptide that is able to bind to the polypyrimidine tract of HAV 5′ UTR. This might result in the curtailment of translation and the promotion of RNA replication.413
Particle Assembly and Release
The mechanisms of HAV particle assembly are likely to differ significantly from those of other picornaviruses, consistent with detection of immature particle intermediates in HAV-infected cells.26,33 Recombinant vaccinia virus–mediated expression of HAV polyprotein or of capsid protein precursor P1-2A, on the one hand, and nonstructural protein precursor 2BC-P3, on the other hand, results in the assembly of
capsid-like structures.60,400 Using this system and C-terminally truncated P1-2A polypeptides, the presence of nonstructural protein 2A as a C-terminal extension of P1 was shown to be essential for both efficient 3C-mediated internal processing of P1-2A and the first step in virion morphogenesis.60 The N-terminal domain of 2A is actually instrumental for assembling five copies of 1AB (VP4-VP2 or VP0), 1C (VP3), and 1D-2A (VP1-2A) polypeptides into pentamers, as a direct primary signal and/or as a consequence of its critical role in 3C-mediated processing of P1-2A, notably at the VP0/VP3 junction. However, whether pentamers are first assembled with five copies of uncleaved P1-2A precursors or cleaved VP0, VP3, VP1-2A polypeptides remains uncertain33,60,300 (Fig. 19.8E). In other picornaviruses, the assembly of pentamers is initiated by N-terminally myristoylated VP4 protein.284 VP4 of HAV, however, is considerably smaller than VP4 proteins of other picornaviruses (at most 23 amino acids) and is not myristoylated, despite the presence of an internal consensus myristoylation signal.367 Twelve pentamers subsequently assemble into complete procapsids (empty capsids) or incorporate virion RNA to form provirions. N-terminal fusion of heterologous sequences to VP0 did not prevent the assembly of procapsids,416 further suggesting that VP4 is not a critical determinant of HAV capsid assembly. The presence of the 3ABC precursor of 3C appears to improve P1-2A cleavage and particle assembly.210 Cleavage at the VP1/2A junction begins after assembly of complete capsids and releases the mature VP1 capsid protein as well as larger VP1 polypeptides of intermediate lengths that, along with uncleaved VP1-2A, can still be detected in low amounts in procapsids and provirions.33,60,300 C-terminal residues of 2A are important for efficient cleavage at the VP1/2A junction,60 which is likely carried out by a cellular protease that remains to be identified. Maturation cleavage of VP0 to release VP2 depends on RNA encapsidation, occurs by an as yet undetermined mechanism, and is associated with a conformational change to generate mature virions.26 Cleavage of VP1-2A and VP0 into corresponding mature capsid proteins is accompanied by an increase in the specific infectivity of viral particles.26,60 The fate of 2A and VP4 in this process is unknown, and efforts to detect them in infected cells or virions have been unsuccessful.
capsid-like structures.60,400 Using this system and C-terminally truncated P1-2A polypeptides, the presence of nonstructural protein 2A as a C-terminal extension of P1 was shown to be essential for both efficient 3C-mediated internal processing of P1-2A and the first step in virion morphogenesis.60 The N-terminal domain of 2A is actually instrumental for assembling five copies of 1AB (VP4-VP2 or VP0), 1C (VP3), and 1D-2A (VP1-2A) polypeptides into pentamers, as a direct primary signal and/or as a consequence of its critical role in 3C-mediated processing of P1-2A, notably at the VP0/VP3 junction. However, whether pentamers are first assembled with five copies of uncleaved P1-2A precursors or cleaved VP0, VP3, VP1-2A polypeptides remains uncertain33,60,300 (Fig. 19.8E). In other picornaviruses, the assembly of pentamers is initiated by N-terminally myristoylated VP4 protein.284 VP4 of HAV, however, is considerably smaller than VP4 proteins of other picornaviruses (at most 23 amino acids) and is not myristoylated, despite the presence of an internal consensus myristoylation signal.367 Twelve pentamers subsequently assemble into complete procapsids (empty capsids) or incorporate virion RNA to form provirions. N-terminal fusion of heterologous sequences to VP0 did not prevent the assembly of procapsids,416 further suggesting that VP4 is not a critical determinant of HAV capsid assembly. The presence of the 3ABC precursor of 3C appears to improve P1-2A cleavage and particle assembly.210 Cleavage at the VP1/2A junction begins after assembly of complete capsids and releases the mature VP1 capsid protein as well as larger VP1 polypeptides of intermediate lengths that, along with uncleaved VP1-2A, can still be detected in low amounts in procapsids and provirions.33,60,300 C-terminal residues of 2A are important for efficient cleavage at the VP1/2A junction,60 which is likely carried out by a cellular protease that remains to be identified. Maturation cleavage of VP0 to release VP2 depends on RNA encapsidation, occurs by an as yet undetermined mechanism, and is associated with a conformational change to generate mature virions.26 Cleavage of VP1-2A and VP0 into corresponding mature capsid proteins is accompanied by an increase in the specific infectivity of viral particles.26,60 The fate of 2A and VP4 in this process is unknown, and efforts to detect them in infected cells or virions have been unsuccessful.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


