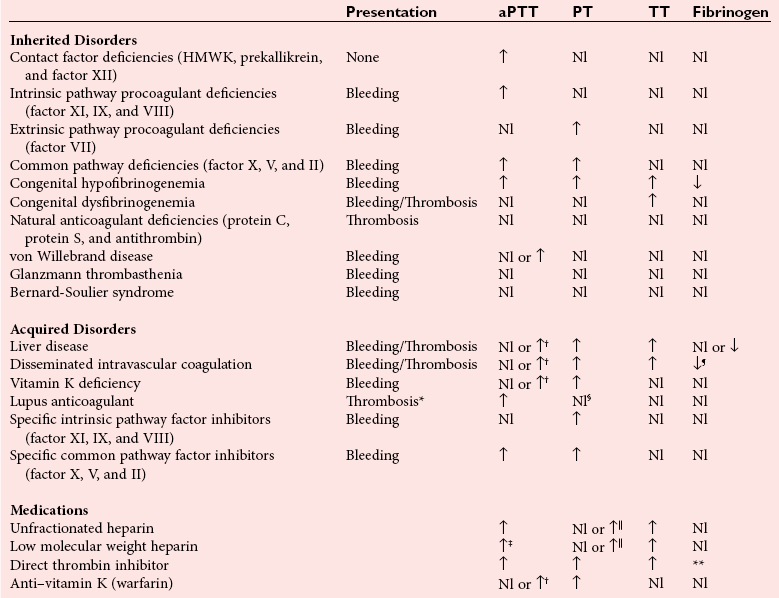
Chapter 59 At the time of an injury, small and medium vessels contract in response to mechanical, neural, and chemical (platelet secretory products) stimuli, in a remarkably successful effort to prevent blood loss. In addition, the endothelial cell plays a key role in the procoagulant and anticoagulant regulation of platelet function and plasma coagulation. Endothelial cells support platelet function through the synthesis and secretion of von Willebrand factor (VWF) and down regulates function by secretion of the potent inhibitor, prostacyclin (PgI2). Anticoagulant functions occurring at the endothelial surface include (1) binding of thrombin to thrombomodulin (TM); (2) conversion of protein C to activated protein C; and (3) clearance of activated coagulation factors by antithrombin (AT). The endothelium also contributes to activation and regulation of fibrinolysis by secretion of tissue plasminogen activator (tPA) and plasminogen activator inhibitors.93 Tissue factor (TF) is found in many tissues in the body, but of particular interest is its presence in the vascular smooth muscle and pericytes that surround the vessel. This vascular distribution of TF has been referred to as the hemostatic envelope.34,39 Figure 59-1 depicts the procoagulant and regulatory roles of the endothelium in hemostasis. Figure 59-1 Role of the Endothelium in Hemostasis. Platelets are key cellular elements in the hemostatic process.77 They are anuclear fragments of the megakaryocyte, produced in the bone marrow and released into the bone marrow sinusoids. They normally circulate for approximately ten days. The functions of the platelet include (Table 59-1): (1). adhesion of the platelet to surfaces at the site of injury—the platelet recognizes non-endothelial surfaces and adheres to those surfaces in an initial step in the hemostatic process; (2). aggregation of platelets at the site of an injury—platelets recognize one another and aggregate, building a structure of platelets at the site of injury; (3) secretion of the two types of granules, alpha granules (contain such proteins as fibrinogen and von Willebrand factor) and dense bodies (contain non-protein components like epinephrine, adenine nucleotides, serotonin and others)—upon platelet activation, granule contents are secreted into the microenvironment, contributing to vascular smooth muscle contraction, further platelet activation and support of plasma coagulation; (4) support of plasma coagulation at the site of the injury—macromolecular enzyme complexes are assembled on the surface of the platelet, greatly accelerating the production of thrombin and, consequently, fibrin; (5) clot retraction may be thought of as the in vivo suture—the platelet contains contractile proteins that shrink the clot by drawing the edges of small injuries closer together, a function that may be an early step in the healing process; (6) support of the injured endothelial cell occurs with the fusion of platelets to damaged endothelium—the platelet becomes a part of the endothelium, helping to support its function.5 Platelet function testing parallels these activities of the platelet. TABLE 59-1 The process of coagulation has three pathways: extrinsic, intrinsic, and common. The extrinsic pathway (also known as the tissue factor pathway) is initiated by release of TF and F is measured by the prothrombin time (PT). The intrinsic pathway (also known as the contact activation pathway) is initiated by activation of the “contact factors” of plasma and is measured by the activated partial thromboplastin time (aPTT) test. The common pathway results from merging of the extrinsic and intrinsic pathways and includes the final steps before a clot is formed. The individual pathways and their interactions are shown in Figure 59-2 and will be discussed later in the chapter. The process of coagulation in vivo proceeds through stages of initiation, amplification, and propagation (Figure 59-3). The initial action of TF and factor VIIa generates small amounts of thrombin, which serve to amplify the reaction through (1) activation of platelets, (2) activation of platelet membrane-bound factors V and VIII, and (3) assembly of macromolecular enzyme complexes on the platelet surface. This leads to amplified generation of thrombin that will feed back to activate factors XI, VIII, and V, propagating the reaction.84 Thrombin plays two key roles in these final steps in the formation of fibrin. First, it is the procoagulant enzyme that cleaves fibrinogen, forming fibrin monomer; second, following fibrin monomer polymerization, thrombin activates factor XIII. Activated factor XIII is a transglutaminase that cross-links the polymer via formation of bonds between lysine and glutamine near the carboxyl end of the γγ chains (within the strands) and bonding between the αγ chains. It is important to keep the process localized to the site of injury, a process that involves regulation of reactions at the site, as well as inactivation of any circulating activated factors on the endothelial surface in the downstream capillary bed. Localization of the process is facilitated by the platelet serving as the template for reactions and by the presence of natural inhibitors like tissue factor pathway inhibitor that neutralize excess enzyme.40,106 Thrombin that is released also binds to TM in the endothelial cell membrane in the capillary bed and activates protein C, which, with cofactor protein S, inhibits excess enzyme generation through inactivation of factors Va and VIIIa. As fibrin is formed at the site of injury, platelets contract, drawing fibrin strands closer together and helping to stabilize the clot and juxtaposing edges of the wound in anticipation of healing. Note: The in vitro (Figure 59-2) and the in vivo (Figure 59-3) coagulation mechanisms are not identical. In vitro, high molecular weight kininogen, prekallikreln and factor XII play a key role in coagulation activation, while these same factors play no role in coagulation in vivo. Figure 59-3 Model of the In Vivo Activation of Coagulation. Fibrinolysis is the process of converting insoluble fibrin to soluble products that are then cleared from the circulation by the liver. The zymogen plasminogen, which is closely associated with fibrin in the clot via molecular sites of recognition, is converted to the enzyme plasmin by urokinase or tPA (Figure 59-4). Plasmin cleaves fibrin to form fibrin degradation products, most commonly measured in the plasma as D-dimer.11,25A Figure 59-4 Activation of Plasminogen and Fibrinolysis. Primary hemostasis is characterized by vascular contraction, platelet adhesion, and formation of a platelet thrombus. It begins immediately after endothelial disruption with platelets being a key cellular component in the hemostatic process.77 Platelets are anuclear fragments of the megakaryocyte cytoplasm, produced in the bone marrow and released into the bone marrow sinusoids. They normally circulate for approximately 10 days. Functions of the platelet are listed in Table 59-1.63 Platelet function testing parallels these activities of the platelet. Automated methods are effective in counting normal numbers of platelets in a background of normal blood cells. Initial platelet counts are nearly always performed using automated methods.10 The specimen that is used is whole blood that has been collected into a tube containing ethylenediaminetetraacetic acid (EDTA). In addition to preventing coagulation, the high affinity of EDTA for calcium inhibits platelet activation. Automated cell counters rely on two basic methods to count platelets in whole blood, both of which exploit the small size of the cell in comparison with other normal cells. In the first of these methods, diluted cells are aspirated through a small aperture across which there is an electrical potential. The lipid envelope of the cell causes a change in the potential when it passes the aperture. Thus, the cell is counted by measuring this change in potential, with the magnitude of the change being proportional to the volume of the cell. Platelets are distinguished from other cells on the basis of their small size. For samples with high platelet counts, statistical adjustments are made to the raw count to adjust for the coincidence of more than one cell passing the aperture simultaneously. The alpha secretory granule, the most common organelle of the platelet, contains a number of proteins (von Willebrand factor, fibrinogen, and many others) that are secreted when the platelet is activated. These granules (proteins) are a deep purple color when stained with Wright-Giemsa stain and give the platelets their delicate granular structure when a routine blood film is examined. Figure 59-5 demonstrates the light microscopic appearance of alpha granules. Figure 59-5 Platelet Morphology. The dense secretory granule of the platelet contains nonprotein molecules (adenine and guanine nucleotides, vasoactive amines, and many others) that are secreted upon activation. These granules have a high content of calcium ion, making them electron dense (see Figure 59-5). Platelets in platelet-rich plasma adhere when placed on a transmission electron microscopy (TEM) grid, where they are then visualized using TEM.144 The method, called whole mount TEM, is rapid and reliably demonstrates the 5 to 10 delta granules seen normally in platelets. Figure 59-5 demonstrates whole mount and thin-section electron microscopy of a normal platelet. Thin-section TEM, although used less frequently than whole mount TEM, provides greater detail of platelet structure and may be helpful in determining the cause of hemorrhage related to abnormal platelet activity.143 Over the past century, many iterations of bleeding time have been employed. Currently, the test is performed on the volar aspect (the surface of the arm on the same side as the palm) of the forearm. A blood pressure cuff is applied at 40 mm Hg to provide uniform intravascular pressure at the site of the incision. A small incision (≈1 cm long and 1 mm deep) is made using a disposable template that provides a uniform incision from test to test. The result is influenced by the direction of incision, with a shorter time obtained if the incision is parallel to the sides of the forearm compared with a perpendicular incision. Blood at the site of injury is gently blotted with a filter paper every 30 seconds until no blood is detectable on the paper (time required ≈4 to 8 minutes). The test is not very sensitive, exhibits high intertechnologist imprecision, and has fallen out of favor, being replaced with other global tests of platelet function like the PFA-100. This may be appropriate for testing of platelet function; but, in most laboratories, bleeding time remains the only method by which to assess the vascular contribution to hemostasis and may continue to be useful for this purpose.19,82 In the United States, many institutions no longer perform the test. Platelets exposed to shear stress, such as occurs at the site of injury, are activated. This characteristic is exploited in the PFA-100 analyzer, a global test of platelet function. The analyzer utilizes a whole blood specimen containing 109 mmol/L trisodium citrate as an anticoagulant. After blood is drawn through a capillary tube, platelets pass through a small perforation in a membrane with embedded activators [collagen/epinephrine and collagen/adenosine diphosphate (ADP)]. The time required for adhering and aggregating platelets to close the perforation is measured (the closure time) (Figure 59-6). The collagen/epinephrine test is sensitive, and if it is found to be normal, the collagen/ADP test usually is not performed. If the collagen/epinephrine test is abnormal, it is followed by the collagen/ADP test. If the collagen/ADP test is normal, the problem is likely related to aspirin or another medication that affects platelet function. If the collagen/ADP test is abnormal, the problem is likely related to an inherited or acquired disorder of platelet function (Table 59-2). The test is affected by anemia and thrombocytopenia (any disorder in which there is an abnormally low number of platelets) and is not performed if the patient has either or both of those conditions. Similar to the test for bleeding time, the test is not sensitive.54 The PFA-200 also provides a method to evaluate the effect of the platelet function-inhibiting thienopyridine drugs on platelets. TABLE 59-2 Expected Results of the PFA-100 Analyzer PFA-100 Platelet Function Analyzer, Siemens Healthcare Diagnostics, Deerfield, Ill. Figure 59-6 Schematic of the PFA-100 Analyzer. Ristocetin is an antibiotic that was originally derived from cultures of Norcardia lurida. It was found to cause in vivo and in vitro aggregation of platelets. Aggregation induced by ristocetin is mediated by VWF. When aggregation is induced by ristocetin, the test will reflect both the activity of VWF in the plasma and function of the receptor for VWF in the platelet membrane (GP Ib-V-IX complex). The methods used to detect aggregation of the platelets are the same as those described in the section on platelet aggregation (see later). Testing is done at a high concentration of ristocetin to detect VWF ristocetin cofactor (VWF:RCo) activity, and at low concentration to detect possible increased affinity of VWF:RCo for its receptor on the platelet. The value of this is discussed later.45,79 The most common of the assays for VWF, it exploits the binding of VWF to platelets in the presence of ristocetin.99 Platelets are prepared free of VWF using gel filtration or washing. Platelets are used fresh for the assay or, as is done for commercial assays, are lyophilized for longer storage and then are reconstituted with a buffer for the assay. Platelets are used in an agglutination reaction with ristocetin as the agonist. The specimen used is platelet-poor plasma (PPP) harvested from whole blood anticoagulated with 109 mmol/L trisodium citrate. Using dilutions of normal pooled plasma, a calibration curve is developed based on the rate or extent of agglutination. The rate or extent of agglutination of the patient’s specimen is then compared with the calibration curve, and the value extrapolated. This original method for determining the activity of VWF is very labor intense, but despite difficulty with imprecision, it remains the gold standard for the development and validation of other methods. The binding of VWF to collagen matrix initiates the process of platelet adhesion and the hemostatic process. VWF activity is quantified using the collagen-binding function.43 The specimen used is PPP harvested from whole blood containing 109 mmol/L trisodium citrate as an anticoagulant. The methods available are primarily enzyme-linked immunosorbent assay (ELISA)-type assays in which VWF binds to stationary phase collagen (collagen types I and III—alone or in combination—derived from human, equine, or bovine tendon), followed by quantification of the bound VWF. The concentration of VWF:CB tends to parallel that of VWF:RCo in most settings. Some assays of VWF:CB are more sensitive to high molecular weight VWF multimers and may detect type 2 VWD (see later) more readily. Monoclonal antibodies to functional epitopes in the VWF (e.g., GP Ib-binding site) have been developed. A homogeneous immunoturbidometric assay with antibody-coated latex beads shows a direct correlation of the degree of agglutination with VWF activity in the plasma.122 These assays have been correlated with other functional assays of VWF. For additional details regarding this type of immunoassay, see Chapter 16. The specimen used is PPP harvested from whole blood anticoagulated with 109 mmol/L trisodium citrate. The original assay for VWF:Ag was the immunoelectrophoretic method of Laurell (referred to as Laurell rocket assay).81 With this method, polyclonal monospecific antibody to VWF is uniformly distributed in a porous agarose gel. Controls and patient specimens are electrophoresed into the gel, the distance of a precipitin “rocket” from the sample well being proportional to the quantity of VWF protein. This method is labor-intensive and time-consuming and has been replaced by classic ELISA assays (see Chapter 16 for description of enzyme-linked immunosorbent and related assays). To simplify the assay and allow for easier automation, currently popular microparticle agglutination assays were developed and are most commonly used. Monoclonal antibody to VWF is coated on microparticles In megakaryocytes and endothelial cells, the VWF gene codes for a subunit protein of approximately 220 kDa. Post-ribosomally, carbohydrate is added to the subunit, dimers of the subunits are formed, and the dimers undergo polymerization. The VWF secreted is a population of molecules varying in size from 500 kDa to as large as 20,000 kDa or more. The size of these VWF multimers is controlled in the plasma by a metalloprotease, ADAMTS13 (see later), the size of the multimers being related to the function of the molecule. These multimers are qualitatively detected in the plasma using electrophoresis in a porous gel, followed by visualization with a radiolabeled or enzymatically labeled antibody to VWF.42,117 Analysis of the multimer patterns is subjective (Figure 59-7). The specimen used is PPP harvested from whole blood containing 109 mmol/L trisodium citrate as an anticoagulant. Loss of the high molecular weight multimers is associated with reduced function (type 2A or 2B VWD), as described later. Figure 59-7 Multimeric Analysis of VWF-Ag. ADAMTS13 is a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13. It also is known as the VWF-cleaving protease and is the enzyme that depolymerizes VWF after secretion from the endothelial cell. When ADAMTS13 is absent or inhibited by autoantibody, thrombotic thrombocytopenic purpura (TTP) can result. The available functional assay for this molecule is based on the ability of the patient’s plasma to cleave the high molecular weight multimers of VWF. It requires lengthy incubation followed by VWF multimer analysis, taking up to 2 days. TTP is a condition of clinical urgency; the current assay is not timely enough for diagnostic purposes, being used only for confirmation after treatment has begun. New assays have become available, including the fluorescence resonance energy transfer (FRET) assay74 and the ELISA,146 which are proving to be of greater diagnostic usefulness. Platelet aggregation and secretion is evaluated by using aggregation and secretion studies. The original platelet aggregation studies were described more than 40 years ago. In the presence of agonist, platelets will stick to each other in a reaction that is mediated primarily by fibrinogen. After more than 45 years, light transmission aggregometry (LTA), with minimal modification, remains the “gold standard” method for evaluating platelet aggregation.12 For LTA, the specimen used is platelet-rich plasma (PRP) harvested from whole blood containing 109 mmol/L trisodium citrate as an anticoagulant. It is also necessary to harvest PPP because the limit of detection of the aggregometer is adjusted with the patient’s own PPP for maximum light transmission, and PRP for minimum light transmission. LTA is a simple turbidometric measurement, the PRP is placed in the light path (37 °C, continuous stirring), and, after the agonist is added, increased transmission of light is recorded over time as the platelets aggregate (Figure 59-8). An alternative method has been developed that allows the use of anticoagulated whole blood. In this case, an electric probe is placed in the specimen and an alternating current passed through the blood. Upon activation, platelets are attracted to the electrodes to which they attach. As more platelets attach, the flow of current is impeded—a change that is plotted over time. Although LTA remains the “gold standard” for platelet aggregation, advantages of the impedance method include (1) testing in the presence of other blood cells, (2) reduced artifact from preparation, and (3) a smaller sample size requirement.89 Regardless of the method used to measure the end point, the agonists used to assess aggregation function are the same. The commonly used agonists are summarized in Table 59-3. TABLE 59-3 Commonly Used Platelet Aggregation Agonists ADP, Adenosine diphosphate; TRAP, thrombin receptor activation peptide. Figure 59-8 Platelet Aggregation. Platelets secrete a wide variety of products into the microenvironment following activation. These products are stored in the platelet delta granules (dense bodies) and alpha granules (Table 59-4). Initial evaluation of platelet secretory function requires a morphologic evaluation (see earlier) to determine whether these granules are present and morphologically adequate. The function of secretion was originally evaluated by exploiting the ability of platelets to take up radioactively labeled serotonin in the laboratory and then measuring release of the label following stimulation of the platelets. The method is very labor-intensive and requires the use of radioisotopes. However it remains the method of choice for evaluation of heparin-induced thrombocytopenia (see later). This method has been replaced in all other clinical settings with the use of the lumi-aggregometer, in which luciferin and luciferase are added to the patient specimen before the agonist is added.145 Conversion of luciferin to its product by luciferase is dependent on adenosine triphosphate (ATP), the only source of which is release from the platelets. In the conversion of luciferin to its product, photons are emitted and are detected by a fluorometer in a quantity that correlates with the ATP released. This method allows for the simultaneous assessment of aggregation and secretion in the same reaction. The method is conducted using LTA or impedance aggregometry. In addition to in vitro demonstration of secretion, surrogate markers of in vivo release can be detected in the plasma. Platelet factor 4 and beta-thromboglobulin are products that are unique to the platelet; when elevated in the plasma, they provide evidence of in vivo secretion. Immunochemical assays (see Chapter 16) are available for both of these analytes. When normal whole blood is allowed to clot in a nonsiliconized glass tube, the combined function of platelets and plasma coagulation will efficiently convert nearly all of the available prothrombin to thrombin (prothrombin consumption). Abnormality of any of the coagulation factors or platelets will reduce the efficiency of this process, and some prothrombin will be left in the serum. This prothrombin is then quantified and compared with the starting plasma prothrombin, allowing an assessment of the percentage of prothrombin that is consumed. If the patient’s prothrombin time and activated partial thromboplastin time are normal, prothrombin consumption reflects the contribution of the platelet to fibrin generation. The test suffers from lack of reproducibility and is not in widespread use, but it can provide information on platelet support of plasma coagulation. It is most frequently abnormal in acquired platelet function disorders such as paraproteinemias and uremia.13 Platelet contractile function is evaluated by determining clot retraction. Upon activation, platelets form filapodia, slender projections from the surface of the cell that attach to fibrin strands. Platelet actin tethers to membrane receptors and organizes with cytoplasmic myosin within these filapodia, forming a rudimentary muscle that contracts at the conclusion of the hemostatic process. This has been described as the in vivo suture, drawing the edges of the injury closer together as healing is initiated. This phenomenon is seen (and is measured qualitatively) by observing the formation of serum in a tube without anticoagulant; the clot shrinks (an active process that depends on the platelet), expressing the serum. This clot retraction is dependent on the contractile function of the platelet, and methods have been described to quantify it. Although not widely used, the technique remains useful, particularly if the more specific information provided by platelet aggregometry is not available.13,125 Flow cytometry is a useful adjunct in evaluating platelet function in selected situations. Agonist receptors and other membrane proteins are detected on the resting platelet. Study of a specific receptor to determine whether it is present and to assess its surface density is used to identify some of the inherited platelet function disorders. Upon activation, the platelet expresses new antigens on the surface or modifies existing receptors. Detecting these modifications is helpful in documenting activation or lack thereof in patients with suspected platelet function disorders. Detection of these surface or activation antigens has been made possible by the development of specific monoclonal antibodies that are labeled with a fluorophor, enabling flow cytometric analysis.92 Verify Now (Accumetrics, San Diego, Calif) is a rapid turbidometric assay that exploits the affinity of activated platelets for fibrinogen. With it, a whole blood specimen is pipetted into a disposable cartridge containing fibrinogen-coated microparticles. By using cartridges with different agonists, the effect of antiplatelet medication is assessed: (1) for glycoprotein IIb/IIIa (GP IIb/IIIa) antagonist drugs, (2) for activation with thrombin receptor activation peptide (TRAP); (3) for aspirin, activation with arachidonic acid; and (4) for thienopyridine drugs, activation with ADP.88 The specimen used is whole blood containing 109 mmol/L trisodium citrate as an anticoagulant, collected into a special plastic 2 mL vacutainer tube. Currently, this method is used only for evaluation of medication effect.76 The “gold standard” for medication effect on platelet function is platelet aggregation using LTA or impedance with the appropriate agonist for the medication (ADP for the thienopyridines; arachidonic acid or collagen for aspirin). Other methods can be compared against this reference method. LTA and impedance are cumbersome, labor-intense methods that do not lend themselves well to urgent or routine testing, leading to the development of Verify Now and PFA-100. Of interest, agreement is still lacking regarding the appropriate concentrations of agonists to be used when evaluating medication effects.76 VWD is the most common of the inherited bleeding disorders. Its exact frequency is not accurately defined. Some have estimated the prevalence to be as high as 1 : 100; however, in clinical practice it does not appear that high. This is a disorder of platelet function that is caused by the absence or abnormality of a plasma protein, VWF. The function of the platelet itself is normal. A second important role for VWF is the binding and stabilizing of coagulation factor VIII in the circulation. The VWF gene, residing in chromosome 12p13.2, is approximately 178 kilobases with 52 exons. The product of the gene is a protein of 2813 amino acids. There is a 22 amino acid pre-peptide and a larger 741 amino acid pro-peptide, both of which are cleaved from the molecule, leaving the mature VWF (2050 amino acids). VWF contains four types of repeated molecular domains.50 Figure 59-9 shows a schematic of the VWF domains and their related functional epitopes. Currently, 526 mutations have been catalogued (http://www.vwf.group.shef.ac.uk/). Analysis of the mutations associated with VWD was initially undertaken to understand the structure and function of the VWF gene, the mRNA, and VWF protein; however, some mutation analysis has now become a part of clinical diagnosis. Details of the mutations are beyond the scope of this chapter. For more information, the reader is referred to Sadler and Blinder.119 The classification of VWD is defined by (1) the quantity of VWF, (2) the relationship of the VWF activity : antigen ratio, (3) the concentration of factor VIII, and (4) the multimeric structure of the VWF:Ag.120 Types 1 and 3 represent quantitative reductions in the factor, and type 2 (2A, 2B, 2M, 2N) represents dysfunctional factor. Table 59-5 summarizes the classification. TABLE 59-5 Classification of Von Willebrand Disease (VWD) Figure 59-9 VWF Protein. Type 3 VWD is the most severe form of the disorder and the least common, occurring in approximately 0.5 to 5 : 1,000,000 in the population. VWF activity and antigen are usually undetectable, and factor VIII is commonly 0.05 U/mL or less. Patients manifest a severe bleeding disorder and often require treatment with VWF-containing products. Multimeric analysis is not performed because VWAg is essentially absent. The condition is inherited as a recessive trait in which patients are homozygous or doubly heterozygous. Mutations, mostly deletions, do not occur in a localized portion of the gene, but are distributed between exons 3 and 52. Because VWF is essentially absent, the amount of factor VIII will also be severely reduced, because factor VIII needs VWF to stabilize it in the circulation. As in all types of VWD, management is focused on replacement of VWF; however, platelet transfusion may also play a role. Alpha granules of platelets of patients with type 3 VWD contain no VWF, the secretion of which is important in adhesion. Thus, in contrast to all other types of VWD, these patients may benefit from platelet transfusion.120 Type 2 VWD occurs as the result of an abnormal or dysfunctional VWF and, as a group, accounts for 20 to 30% of cases of VWD. As might be expected, with a multifunctional, large protein, mutations have led to a number of variations in the dysfunction of the molecule. The classification requires analysis of the VWF multimers because the loss of high molecular weight multimers is associated with dysfunction in two of the subtypes of VWD—2A and 2B. Current subtypes of the dysfunction of the molecule are listed in Table 59-5, and each are discussed below. Type 2 VWD is suspected when there is a discrepancy between VWF activity and the VWF antigen. Ratios of Type 2A VWD is associated with abnormal multimer analysis and loss of high molecular weight multimers. It is the most common of the type 2 subtypes, accounting for 10 to 15% of cases of VWD. High molecular weight multimers are critical for the platelet adhesion event, and their loss reduces adhesion, leading to a mild to moderate bleeding diathesis. Molecular lesions are most frequently identified in the exon 28 region of the gene (the A2 domain); however, mutations have been described in other domains. Inheritance is generally dominant, especially if the mutation is located in the A2 domain. If inheritance is recessive, there may be value in analyzing other domains of the gene. The severity of the bleeding diathesis tends to be related to VWF:RCo activity in the plasma. As with type 2M, treatment of type 2A VWD with desamino-D-arginine vasopressin (DDAVP) is variable, and management generally requires the infusion of VWF-containing products. Platelet transfusion is not recommended.120 Type 2B VWD is associated with loss of the high molecular weight multimers of VWF:Ag; however, the proportion of those multimers that are missing is often less than that seen in type 2A. Type 2B VWD is uncommon, accounting for less than 5% of cases of VWD. The mutations that have been described are also in exon 28 (the A2 domain), at or near the GP Ib binding epitope of the mature protein. The abnormality gives the VWF protein a higher affinity for the GP Ib receptor on the platelet surface. This increased affinity is detected in the laboratory by demonstrating RIPA at a low concentration of ristocetin (usually ≤0.5 mg/mL)—a concentration that will not induce aggregation in the presence of normal VWF. This testing is indicated whenever multimeric analysis shows loss of the high molecular weight VWF:Ag multimers and is the method used to distinguish type 2A from type 2B VWD. In type 2B VWD, binding of VWF to platelets in vivo may occur spontaneously or with minimal stimulus, causing in vivo aggregation and clearance of the platelets. Thus, thrombocytopenia is a common finding in patients with this disorder, and consumption of VWF in this manner contributes to decreased amounts of VWF. Patients with type 2B VWD present with a mild to moderate bleeding diathesis. Management is generally limited to infusion of VWF-containing products. Use of DDAVP is not recommended because of the possibility that the release of VWF may induce in vivo platelet agglutination and precipitate a thrombotic complication. Platelet transfusion is not recommended.120 Platelet-type VWD (pseudo-VWD) is a syndrome with reduced high molecular weight multimers due to increased affinity in the receptor for VWF on the platelet. VWD type 2B and platelet-type VWD are difficult to distinguish clinically. See the discussion of the Bernard Soulier syndrome, later. Type 2N VWD results in a decrease in plasma factor VIII but normal VWF-mediated platelet function. Thus, the phenotype of the disorder is similar to that of hemophilia but shows autosomal inheritance. All testing of VWF functions and antigen is normal. Mutations in the VWF gene occur in exons 20 to 28 (the D′ domain), which code for the epitope in the VWF protein that binds to factor VIII. A few specialty laboratories are able to measure the binding of factor VIII to patient VWF, confirming the diagnosis. Distinguishing type 2N VWD from hemophilia is important because treatment of the disorders requires the infusion of different products.120 Type 1 VWD is the most prevalent form of VWD, accounting for approximately 70 to 80% of cases.101 Inheritance of type 1 VWD is autosomal; however, the phenotype of the associated bleeding tendency is quite variable, with many patients manifesting a mild to moderate bleeding disorder, while others with similar laboratory values do not manifest a bleeding tendency. With laboratory analysis, one generally finds a concomitant reduction in VWF activity, VWF antigen, and factor VIII. Values vary from below 0.2 IU/mL to as high as 0.5 IU/mL. Genetic analysis of patients is complex. European and Canadian reports of the molecular analysis of patients with type 1 VWD show that of those patients with clearly normal multimer analysis, only 55 to 63% had an identifiable mutation, and that those mutations were not localized in the gene.21,52 There is a relationship between the concentration of VWF in the plasma and the probability of detecting a mutation in the gene. Of those with 0.15 IU/mL or less VWF protein, 95% had a mutation, while only 50% of those with 0.45 IU/mL or greater VWF protein had mutations. Among those who have mutations, 10 to 15% have more than one mutation identified.50 What of those patients with reduced VWF and no identifiable mutation? Carboxylation of the VWF protein is closely associated with the ABO blood group of normal individuals. Those with blood group O have lower circulating VWF than those who have the A and/or B antigen. It is thought that carboxylation may influence the half-life of the molecule.49 Analysis of the blood groups of patients with type 1 VWD shows a significantly higher number of patients with blood group O. Thus, changes in the postribosomal carboxylation of VWF may account for VWD without mutations in the VWF gene. Management of bleeding in type 1 VWD depends on the severity and expected duration of the hemorrhagic challenge. Those with a mild challenge of short duration can receive DDAVP, while those with more extensive challenge require infusion with VWF-containing products.119 The Bernard-Soulier syndrome (BSS) is a disorder of platelet function that is related to the function of VWF; however, in this condition, the amount of VWF protein is normal, with the lesion being in the VWF receptor on the surface of the platelet, GP Ib/V/IX. The disorder is inherited as an autosomal recessive trait with bleeding seen only in homozygotes, and it is seen most frequently in consanguineous relations. It is a rare disorder, with a prevalence of less than 1 : 1,000,000 in the general population. Mutations have been reported in the genes coding for GP Iba (gene symbol GP1BA) and GP IX (GP9), but not GP V (GP5). For further details, see http://www.bernard-soulier.org/mutations. It is one of the giant platelet syndromes with concurrent thrombocytopenia and is associated with moderate to severe bleeding. Bleeding episodes are managed with platelet transfusions. While discussing mutations of the GP Ib/V/IX receptor, it is useful to mention platelet-type or pseudo-VWD. This is also an inherited bleeding syndrome in which there is a normal VWF gene and protein, but a lesion in the platelet receptor causes an increased affinity with VWF. This disorder is difficult to distinguish from type 2B VWD. The diagnosis relies on demonstration that the VWF is normal in the plasma and that the platelets are abnormal (complex mixing studies are done in some laboratories), or the differential diagnosis is made with mutation analysis. The distinction is important because the condition is managed with platelet transfusion.78 Absence or abnormality of the platelet integrin, GP IIb/IIIa, the receptor for fibrinogen in aggregation, is the cause of Glanzmann thrombasthenia (GT). Mutations (deletions or other mutations) in the genes for GP IIb or GP IIIa membrane proteins have been identified. The disorder is inherited as an autosomal recessive trait that leads to a mild to moderate bleeding diathesis. It is a rare disorder, affecting approximately 1 : 1,000,000 in the general population; however, it may be more prevalent in populations with consanguineous relations. Platelet count is normal; however, platelet aggregation with all agonists except ristocetin is abnormal. In addition, the bleeding time is prolonged, the PFA-100 value is prolonged, and clot retraction is abnormal. Bleeding episodes are treated with platelet transfusion.98 Many medications affect platelet function. For example, drugs such as aspirin, thienopyridines, and monoclonal inhibitors of GP IIb/IIIa are used specifically to reduce function when inhibition of thrombosis, particularly arterial thrombosis, is desired. In other cases, the effects on platelet function are adverse complications of the medication. These effects generally are not enough to lead to a bleeding complication; however, if the medication is used in patients with inherited or acquired platelet disorders or other disorders of hemostasis, the effect may increase bleeding. Examples of medications that affect platelet function are listed in Table 59-6.8 TABLE 59-6 Examples of Medications Affecting Platelet Function In patients with renal failure, organic acids such as guanidinosuccinic and phenolic acids are abnormally retained in the blood. These acids have a deleterious effect on all platelet functions and are often associated with a bleeding diathesis. Neither creatinine nor urea has a significant effect on platelet function, but they provide a useful surrogate marker for other retained products. Platelet functions tend to be affected at urea nitrogen of ≈60 mg/dL or greater and creatinine of 3 to 4 mg/dL. In vitro studies have confirmed the deleterious effects of organic acids and the rapid rate at which normal platelets demonstrate loss of function. For this reason, the use of platelet transfusion in managing hemorrhage in uremic patients is ineffective. Management is difficult; however, treatment with dialysis to reduce retained acids is often effective. In addition, the use of DDAVP has proven of some value.139 Treatment of anemia by keeping the hemoglobin above 10 g/dL using erythropoietin and transfusion has a beneficial effect in managing bleeding.95 Patients with multiple myeloma or Waldenström macroglobulinemia often have remarkably elevated concentrations of immunoglobulins. These immunoglobulins are associated with hemorrhage via two basic mechanisms. First, the paraproteins interfere with fibrin-monomer polymerization, which is reflected in prolonged thrombin time. Second, the proteins adsorb nonspecifically to the surface of the platelet, masking the receptors on the surface of the platelet and reducing platelet activity. These problems are managed by reducing the concentration of the protein through management of the underlying malignancy. In acute situations, plasmapheresis is effective in reducing paraprotein concentrations, but only for short-term management.38 In myeloproliferative and myelodysplasplastic syndromes, the megakaryocyte, similar to other cell lines, is affected by malignant transformation. The effects of malignant megakaryocyte production of platelets on platelet function are widely variable. These effects may include but are not limited to (1) increased or decreased platelet counts, (2) reduced granules, (3) reduced or abnormal membrane receptors, (4) adsorption of high molecular weight VWF multimers, causing an acquired type 2 von Willebrand disease (VWD), or (5) abnormal signal transduction mechanisms. As a result of these variable effects, patients may manifest abnormal bleeding or, paradoxically, thrombosis. Bleeding episodes may be managed with platelet transfusion; however, in patients with thrombocytosis, it is difficult to determine whether the risk to the patient is bleeding or thrombosis. Patients at thrombotic risk may benefit from treatment with antiplatelet medication.109 Control of preanalytical issues in coagulation testing is paramount to good laboratory performance. In addition to the common issues of hemolyzed, icteric, or lipemic samples, some preanalytical factors of particular importance in coagulation testing include (1) clotted specimens, (2) improper blood-to-anticoagulant ratio, and (3) contamination with heparin or other anticoagulants. Traumatic venipuncture, activation of blood within the collection device, or improper mixing of the anticoagulant with blood may result in clotting, which consumes coagulation factors, making testing unreliable. Blood for coagulation testing should be collected by standard venipuncture techniques into 109 mmol/L (3.2%) trisodium citrate, such that the final ratio of blood to anticoagulant is 9 : 1. Blood is commonly collected into commercially available tubes with prealiquoted trisodium citrate and a line indicating the appropriate volume of blood to be drawn. Collection of a volume of specimen less than the recommended volume (“a short draw”) will result in excess anticoagulant and prolonged clotting times. Likewise, samples with high hematocrit (>55%) will require a decreased volume of anticoagulant because of a lower plasma volume. Some coagulation testing, such as activated clotting time, may be performed on whole blood; however, most routine clot-based assays are performed on platelet-poor plasma. To avoid interference from phospholipid and other platelet-derived substances such as platelet factor 4, platelet-poor plasma is prepared by centrifugation, typically at 1500 g for at least 15 minutes. Phospholipids are important for the spatial orientation of coagulation molecules and have a significant impact on the activated partial thromboplastin time. Platelet factor 4 binds heparin and may alter clot-based tests. Centrifugation and testing should occur as soon as possible, usually within 24 hours for prothrombin time (PT), or within 4 hours for aPTT. Centrifuged samples may be stored at 18 to 24 °C, whereas colder temperatures should be avoided because of possible activation of factor VII. When testing is delayed, the plasma should be separated from the cells and kept below −20 °C or at −70 °C for longer storage.25 Household grade freezers with autodefrost cycles are not suitable. As a result of high-volume testing, most coagulation testing is now performed on automated instruments that control the temperature of the reaction and detect end points by use of any one of several methods. Most methods detect a change in physical/mechanical properties or a change in the light transmission produced by polymerized fibrin. Numerous approaches for mechanical end point detection have been developed. One mechanical method consists of a metal ball at the bottom of a sample cuvette that is sent into a back-and-forth motion by a magnet; the end point is detected when fibrin monomers polymerize into fibrin strands and impede the motion of the ball. Another mechanical detection system uses a magnet to hold a ball to the side of a rotating cuvette until fibrin strands physically displace the ball. Optical detection (usually nephelometric but occasionally turbidometric, Chapter 10, Optical Techniques) is the most commonly employed method, detecting a decrease in light transmission or increased light scatter as fibrin monomers are polymerized into fibrin strands.124 Optical end points may occur at preset thresholds; however, other instruments use the kinetics, such as maximum acceleration of fibrin polymerization, to define end points. Light sources have traditionally been halogen lamps or lasers, but newer instruments may use light-emitting diodes (LEDs) that increase longevity and allow measurement at wavelengths that have less overlap with interfering substances. Differences are expected between end point detection systems. An advantage of mechanical over optical end point detection is the ability to measure clots in the presence of substances such as hemoglobin, bilirubin or lipid that interfere with optical methods. As opposed to primary hemostasis, which involves the processes of vascular contraction and platelet function, secondary hemostasis is defined as “the formation of fibrin through the coagulation cascade” (http://ahdc.vet.cornell.edu/clinpath/modules/coags/second.htm). Activity of coagulation factors II, VII, IX, and X and natural anticoagulants, protein C, and protein S, is dependent on the action of vitamin K (see Chapter 31). Vitamin K–dependent carboxylases convert glutamic acid residues to gamma-carboxyglutamic acid residues on vitamin K–dependent coagulation factors, imparting calcium binding function to these factors. Reduced vitamin K is regenerated through the action of a vitamin K epoxide reductase. Deficiencies of vitamin K result in inactive coagulation factors as the result of inadequate gamma carboxylation with resultant loss of calcium binding capability. Vitamin K deficiency, therefore, may lead to an acquired bleeding disorder. Oral vitamin K antagonists (VKAs), such as warfarin, are used for individuals who require sustained anticoagulation. VKA inhibits vitamin K epoxide reductase that is responsible for regenerating the active vitamin K cofactor necessary for gamma carboxylation. Because protein C is a vitamin K–dependent natural anticoagulant with a short half-life, patients treated with VKA alone have an initial risk of thrombosis until the other vitamin K–dependent procoagulants are also reduced. For this reason, anticoagulant effect is first achieved with heparin, and then oral VKA is overlapped with heparin therapy for 3 to 5 days. A study comparing combined VKA and heparin therapy against VKA therapy alone for proximal vein thrombosis was terminated early because of an increased rate of symptomatic extension or recurrence of venous thromboembolism in the group treated with VKA alone.15 The PT is a clot-based assay that reflects the activity of extrinsic and common pathway factors of the coagulation cascade. The PT is initiated by the addition of a thromboplastin reagent containing tissue factor, calcium, and phospholipid. The PT cannot be compared among different laboratories because the specific thromboplastin/instrument combination determines the responsiveness of the test. The INR, which is derived mathematically from the PT, allows harmonization of PT across laboratories for the purpose of monitoring VKA. All results are standardized using the international sensitivity index (ISI) for the particular thromboplastin/instrument combination used to perform the test.127 ISI is based on the slope of the relationship between log PT values obtained with a test thromboplastin reagent and a reference thromboplastin, using samples from normal individuals and patients on VKA. The INR is then calculated as follows: The PT/INR is performed on platelet-poor plasma at 37 °C. The tissue factor in the reagent initiates the extrinsic pathway of the coagulation cascade (see Figure 59-2), the phospholipids provide a surface for assembly of coagulation factors, and the calcium chloride counteracts the binding of plasma calcium by citrate, making calcium available for the coagulation process. The timer is started when reagent is added, and the end point occurs when fibrin monomer polymerization is detected. The PT/INR is used to identify deficiencies or inhibitors of factors VII, X, V, and II and fibrinogen. Expected results of clotting assays, including PT/INR, are shown in Table 59-7 for key inherited and acquired disorders. TABLE 59-7 Clinical Settings and Coagulation Tests *Thrombosis associated with antiphospholipid syndrome. †Prolongation of aPTT depends on the sensitivity of the reagents and the degree of factor deficiency. ‡aPTT is not predictably prolonged by low molecular weight heparin. §Rare lupus anticoagulants prolong PT in addition to aPTT. ||PT reagents may contain heparin-neutralizing reagents (usually up to 1 U/mL). ¶Fibrinogen decreased with overt DIC. **Direct thrombin inhibitors may cause underestimation of fibrinogen by the Clauss method. The sensitivity of PT/INR reagents to the deficiency of coagulation factors varies with the specific reagent and instrument combination. Therefore, it is useful for laboratories to determine the sensitivity of a given PT/INR system to factors VII, X, V, and II. This is accomplished by measuring the PT/INR in a dilutional series of factor-deficient plasma with normal pooled plasma. The activity at which the PT/INR is prolonged beyond the upper limit of the reference interval signifies the sensitivity of the assay to the factor being tested (Figure 59-10; for illustration, this example uses the aPTT test rather than PT/INR). If a reagent is overly sensitive to decreases in a factor (e.g., 0.5 IU/mL factor VII), the laboratory may detect clinically insignificant prolongations of the PT/INR. Needless laboratory evaluations and delays in surgical interventions may be avoided if reagents are selected carefully. Figure 59-10 Relationship of aPTT to Factor VIII Activity. The aPTT is a clot-based assay that is initiated by activation of contact factors and reflects the activity of the intrinsic and common coagulation pathways (see Figure 59-2). Activation of contact factors [high molecular weight kininogen (HMWK), prekallikrein, and factors XII, and XI] is achieved by the addition of one of a wide variety of activators, most having in common a negatively charged surface. Activators including kaolin, celite, and micronized silica have been used extensively when mechanical end points are determined by coagulometers (see earlier); however, if an optical detection is used, micronized silica or the soluble chemical activator, ellagic acid, is used because they do not interfere with light transmission. The aPTT is sensitive to activities of the intrinsic and common pathway factors (1) HMWK, (2) prekallikrein, (3) XII, (4) XI, (5) IX, (6) VIII, (7) X, (8) V, (9) II, and (10) fibrinogen.14 Common uses of the aPTT include monitoring of heparin therapy and screening for deficiencies or inhibitors of intrinsic and common pathway factors. The use of aPTT for monitoring heparin therapy is discussed later. The response of aPTT reagents to (1) heparin,71 (2) factor deficiency, (3) specific factor inhibitors, and (4) lupus anticoagulants16 varies widely. If a clinical laboratory intends to use aPTT to detect factor deficiencies, it is important to know the threshold of factor deficiency that prolongs the aPTT. The response of aPTT to factor VIII and factor IX is particularly important because of the prevalence of hemophilias A and B. Determining this responsiveness is accomplished by measuring the aPTT in a dilutional series of factor-deficient plasma with normal pooled plasma. The activity at which the aPTT is prolonged beyond the upper limit of the reference interval signifies the sensitivity of the assay to the factor being tested (see Figure 59-10). In general, the desired response to factors VIII, IX, and XI should be approximately 0.3 IU/mL. Very sensitive or insensitive reagents may create problems. The aPTT is prolonged in a variety of clinically significant and insignificant scenarios. Table 59-7 summarizes some causes of prolonged aPTT and/or PT/INR. Seven general potential causes of isolated prolongation of aPTT include (1) a procoagulant deficiency that may be associated with a bleeding history—may include multiple factor deficiencies; (2) a contact factor deficiency without bleeding risk (XII, prekallikrein, HMWK); (3) a specific inhibitor acquired as an alloimmune or autoimmune phenomenon (e.g., factor VIII inhibitor); (4) a nonspecific inhibitor such as a lupus anticoagulant; (5) a medication effect or contamination (e.g., heparin, direct thrombin inhibitor); (6) a spurious result; and (7) an extreme of the population in which the aPTT may be minimally increased outside the upper limit of the reference interval, usually defined by two standard deviations, which includes only 95% of the population. Although activated partial thromboplastin time implies the addition of thromboplastin, this is not the case. This ambiguity is a result of use of the term “partial” thromboplastin by Langdell and associates80 to describe a group of reagents that produced clotting that was less rapid in hemophiliac plasma than in normal plasma. This was contrasted with “complete” thromboplastin reagents used in the prothrombin time assay, which did not discriminate normal and hemophiliac plasmas. Partial thromboplastin activity was achieved by replacing thromboplastin with cephalin or diluting thromboplastin; however, the overall effect of the dilutions was to remove the thromboplastin activity, making the contribution of the intrinsic pathway measurable. Subsequently, the addition of activators led to the aPTT that we recognize. aPTT may be prolonged in patients who are not at risk for bleeding, such as those with deficiency of factor XII, prekallikrein, or HMWK. In fact, the intrinsic pathway is best considered a laboratory phenomenon, and in vivo coagulation is better considered in terms of initiation, amplification, and propagation (see Figures 59-2 and 59-3). In vivo, the initiation phase of coagulation is rapidly inhibited by tissue factor pathway inhibitor, with accompanying feedback by thrombin that initiates the propagation phase of coagulation. The aPTT reflects clinically significant deficiencies of factors in the propagation phase (factors XI, IX, and VIII) of in vivo coagulation and is an important test for identifying these deficiencies.
Hemostasis
Vascular
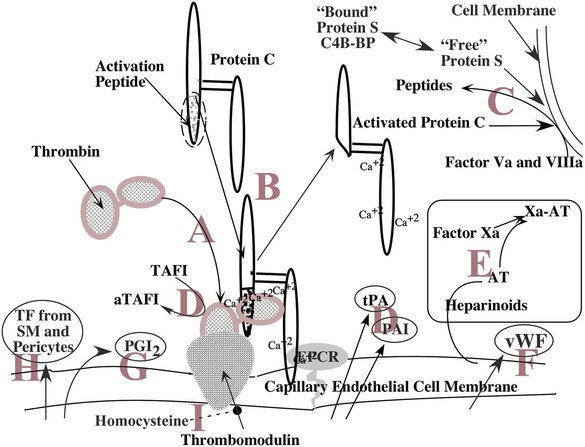
[A] Thrombin released into the circulation at the site of an injury binds to thrombomodulin (TM), an endothelial cell membrane protein. Bound to TM, thrombin does not cleave fibrinogen. [B] Protein C, bound to the endothelium by endothelial cell protein C receptor (EPCR), is cleaved by thrombin bound to thrombomodulin; the activation peptide is removed and activated protein C (APC) released. [C] At a distant site of coagulation, APC with its cofactor, the free form of protein S, cleaves factors Va and VIIIa, inactivating them and downregulating coagulation at the site. [D] Fibrinolysis is also regulated by actions at the surface of the endothelium by the release of tissue plasminogen activator (tPA) and plasminogen activator inhibitors (PAI), as well as the activation of thrombin-activatable fibrinolysis inhibitor by thrombin bound to TM. [E] Antithrombin (AT) associated with heparin-like compounds in the endothelial cell membrane binds to activated serine proteases such as factor Xa, inhibiting their function through covalent linkage, allowing clearance from the circulation by the liver. [F] VWF, a key molecule in platelet adhesion, is synthesized by the endothelial cell and secreted both into the blood and abluminally into the subendothelial basement membrane. [G] Prostaglandin synthesis in the endothelial cells produces PGI2, a potent platelet inhibitor, and secretes it into the blood. [H] Tissue factor (TF) is synthesized in the smooth muscle and pericytes of small vessels forming the hemostatic envelope. [I] Homocysteine in the endothelial cell inhibits the expression of TM in the endothelial cell membrane.
Platelets
Function
Description
Adhesion
Upon activation, the platelet recognizes surfaces other than normal endothelium and adheres to those surfaces.
Aggregation
Upon activation, the platelet recognizes and attaches (aggregates) to other platelets.
Secretion
Upon activation, the platelet secretes the contents of the alpha granules and dense bodies (delta granules).
Support of plasma coagulation
At the site of injury, the platelet serves as a surface upon which macromolecular enzyme complexes form and plasma coagulation is accelerated.
Clot retraction
Following clot formation, the filapodia of platelets attach to the fibrin strands and, through contraction, reduce clot size and express serum in vitro and juxtapose edges of the injury in vivo.
Support of damaged endothelium
Platelets adhere to damaged endothelium, fuse with the membrane, and become incorporated with the endothelial cytoplasm.
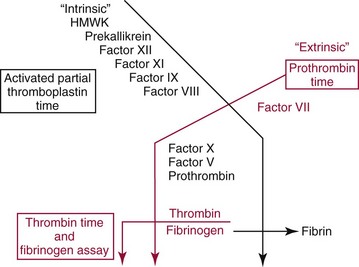
Coagulation
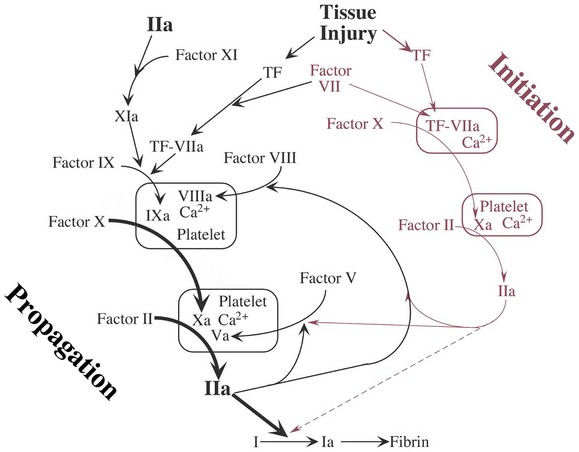
At the site of injury, tissue factor (TF) is released and binds to and activates factor VII. In the presence of calcium (Ca2+), the complex activates factor X. A macromolecular complex is formed on phospholipid (surface of the platelet) with the cofactor, factor V, and initial limited quantities of thrombin (IIa) are produced from prothrombin. This process has been called extrinsic coagulation and is the initiation of the coagulation process. This small amount of thrombin serves to activate platelets at the site, activate factor V, activate factor VIII, and activate factor XI. This feedback activation leads to acceleration of the cascading activation of enzymes on the surface of the activated platelet with a resulting burst of IIa generation and the formation of fibrin. The activities enclosed in the boxes depict the formation of macromolecular enzyme complexes that involve vitamin K–dependent coagulation factors (II, VII, IX, and X) and their cofactors (VIIIa and Va), joining with Ca2+ on the surface of the platelet.
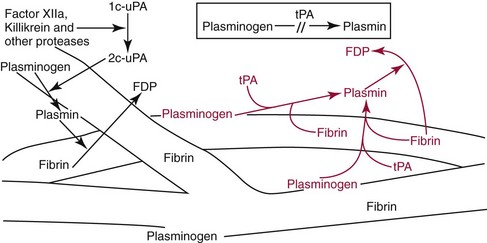
In vivo, plasminogen is activated to the plasmin (a serine protease) by two pathways. First, (shown in black) single-change urokinase (1c-uPA) is converted to active two-chain urokinase (2c-uPA) by contact activation factors and other proteases. Then 2c-uPA cleaves plasminogen to form plasmin. Second, tissue plasminogen activator (tPA) (shown in red) converts plasminogen to plasmin only in the presence of its cofactor, fibrin. Fibrin as a cofactor for this reaction is of interest because it is also the substrate for the enzyme being generated. Without fibrin, conversion of plasminogen to plasmin is too inefficient to be physiologically relevant.
Primary Hemostasis
Evaluating Platelet Number
Automated Platelet Counting
Evaluating Platelet Structure
Evaluating Alpha Granules
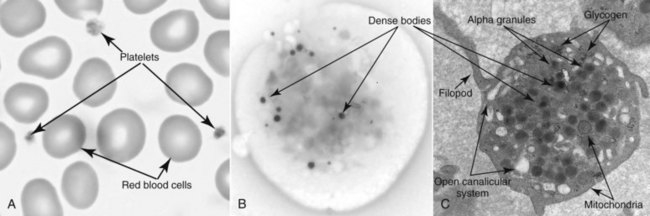
A is a photo of a Wright-Giemsa–stained blood film with several red blood cells and three platelets. Normally, the platelets demonstrate some variation in size and shape, as seen here. Variable staining of the cytoplasm of the platelet is due to the alpha granules, the proteins of which stain deep purple. B is an electron micrograph of a whole mount of a platelet. The purpose of the preparation is to demonstrate the dense bodies (electron dense because of the high concentration of calcium). Visualization of other organelles requires thin-section preparation. C is a thin-section electron micrograph demonstrating many of the platelet organelles.
Evaluating Dense Bodies (Delta Granules)
Evaluating Global Platelet and Vascular Function
Bleeding Time
Platelet Function Analyzer (PFA-100 and PFA-200)
Condition
Collagen/Epinephrine Cartridge Closure Time
Collagen/ADP Cartridge Closure Time
Normal
Normal
Normal
Medication (aspirin effect)
Prolonged
Normal
Platelet function defect
Prolonged
Prolonged
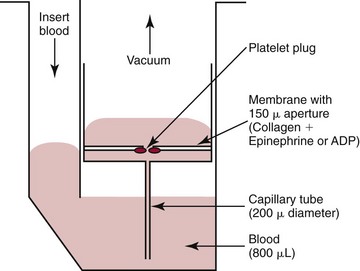
The PFA-100 analyzer provides a global measure of platelet function in a whole blood specimen. Blood anticoagulated with citrate is pipetted into the cuvette with a reaction membrane with collagen/epinephrine or collagen/ADP. Using a vacuum (syringe suction), blood is aspirated through the capillary tube at a rate sufficient to produce a wall shear rate of 1000 seconds−1. Blood containing shear stress–activated platelets enters a small chamber and passes through the aperture of the membrane. At the aperture, the platelets adhere and aggregate until the aperture is closed and flow is obstructed. The time to achieve closure is referred to as closure time.
Evaluating Platelet Adhesion
Ristocetin-Induced Platelet Aggregation Assay
von Willebrand Factor–Ristocetin Cofactor Assay (VWF:RCo)
von Willebrand Factor–Collagen-Binding Assay (VWF:CB)
von Willebrand Factor Immunoactivity Assay (VWF:IA)
von Willebrand Antigen Assays (VWF:Ag)

VWF:Multimer Assay
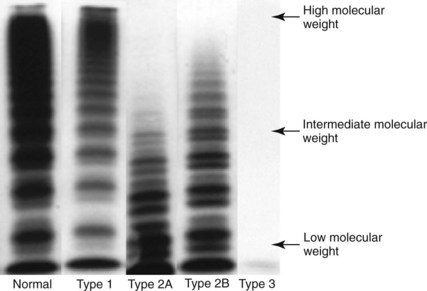
Plasma is electrophoresed into a porous gel of agarose or agarose and acrylamide, the multimers are separated, and the VWF:Ag multimers are radiolabeled with antibody to VWF:Ag. Typical patterns of normal and VWD types 1, 2A, 2B, and 3 are shown in the autoradiograph. The pattern seen in type 2M is like that of type 1. (Image courtesy M. R. Ledford-Kraemer.)
ADAMTS13 Assay
Evaluating Platelet Aggregation and Secretion
Platelet Aggregation Studies
Collagen
Activates receptor GP VI
ADP
Activates receptors P2Y1 and P2Y12
TRAP
Activates the thrombin receptors PAR1 and PAR4
Epinephrine
Activates the alpha 2 receptor
Arachidonic acid
Activates the cyclooxygenase pathway
Ristocetin
Activates VWF binding to GP Ib/V/IX
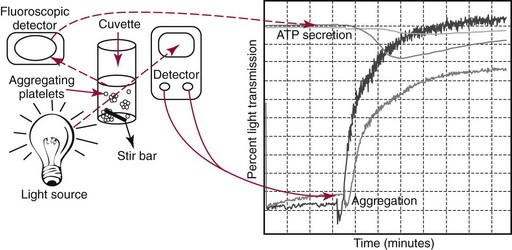
Platelet-rich plasma harvested from whole blood collected in citrate anticoagulant is stirred continuously at a constant rate at 37 °C. Increasing absorbance (loss of turbidity) following addition of an agonist is measured. The gain of the detector is set using the patient’s platelet-rich plasma and platelet-poor plasma. The reaction may be quantified by measuring its rate or extent. Measuring secreted adenosine triphosphate (ATP) reflects secretion of platelet-dense bodies from the platelet. Luciferin and luciferase are added to the aggregation reaction. Luciferase converts luciferin to its product in a reaction that is absolutely dependent on ATP and emits photons. Fluorescence is measured, and secretion is quantified as a function of the quantity of ATP released.
Platelet Secretion
Evaluating Platelet Support of Plasma Coagulation
Evaluating Platelet Contractile Function
Evaluation of Platelets Using Flow Cytometry
Evaluation of Antiplatelet Medication
Verify Now
Platelet Aggregation
Clinical Use of Laboratory Tests for Platelet Disorders
Inherited Disorders
von Willebrand Disease
Type of VWD
Incidence
Description
Type 1
70-80%
Mild to moderate VWD with corresponding reduction of VWF:RCo, VWF:CB, and VWF:Ag, and factor VIII of variable magnitude
Type 3
<1%
Severe VWD with near absence of VWF and factor VIII
Type 2A
10-15%
Dysfunctional VWF with reduced high molecular weight VWF:Ag multimers
Type 2B
5%
Dysfunctional VWF with reduced high molecular weight VWF:Ag multimers and increased affinity of VWF for the GP Ib-IX-V receptor on the platelet
Type 2M
<1%
Dysfunctional VWF with normal distribution of VWF:Ag multimers
Type 2N
<1%
Dysfunctional VWF with reduced binding of factor VIII manifesting as reduced circulating factor VIII and normal platelet function
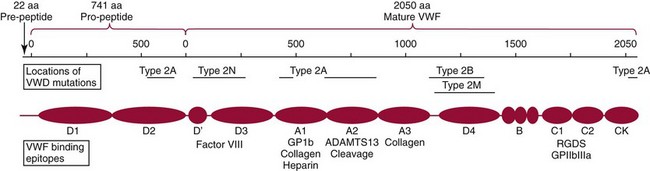
The VWF gene, residing in chromosome 12p13.2, is approximately 178 kilobases with 52 exons. The product of the gene is a protein of 2813 amino acids. There is a 22 amino acid pre-peptide and a larger 741 amino acid pro-peptide, both of which are cleaved from the molecule leaving the mature VWF (2050 amino acids). VWF contains four types of repeated molecular domains. The figure depicts the domains, the locations of functional epitopes, and the locations of the most common mutations in the types of VWD. The protein forms dimers by linkage of the amino terminal end with subsequent formation of multimers via polymerization of the dimers.
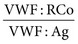 of 0.5 to 0.6 or less indicate a dysfunctional molecule and serve as an indication to test further, as described later, to subtype the disorder. In general, the ratio
of 0.5 to 0.6 or less indicate a dysfunctional molecule and serve as an indication to test further, as described later, to subtype the disorder. In general, the ratio 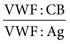 will be lower than the ratio
will be lower than the ratio 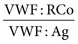 in type 2A and 2B, but the reverse is true in type 2M.120
in type 2A and 2B, but the reverse is true in type 2M.120
Bernard-Soulier Syndrome
Glanzmann Thrombasthenia
Acquired Disorders
Medication
Class of Medication
Examples
Nonsteroidal anti-inflamatory
Aspirin, naproxen, ibuprofen, indomethacin
GP IIb/IIIa antagonists (antibodies)
Abciximab, eptifibatide, tirofiban
Thienopyridines
Clopidogrel, ticlopidine
Increase cAMP and/or cGMP
Iloprost, dipyridamole, prostacyclin
Cardiovascular medications
Many
Volume expanders
Dextran, hydroxyethyl starch
Chemotherapeutic agents
Mitomycin, daunorubicin, BCNU
Psychotropic medications
Many
Others
Antihistamines, clofibrate, rad contrast agents
Renal Failure
Paraproteinemia
Primary Bone Marrow Disorder
General Considerations in Coagulation Testing
Preanalytical Variables for Coagulation-Based Assays
Optical versus Mechanical End Point Detection
Secondary Hemostasis
Vitamin K–Dependent Factors
Evaluation of Secondary Hemostasis
Prothrombin Time/International Normalized Ratio (PT/INR)

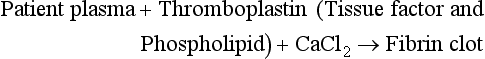
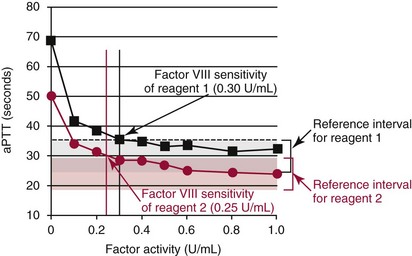
The graph demonstrates typical relationships of factor activity to clotting time. Response curves are shown for two aPTT reagents. Reference intervals for the reagents are represented as gray and red shaded areas for reagent 1 and reagent 2, respectively. The factor activity at which aPTT prolongs above the reference interval is the limit of detection of the reagent (shown as black or red arrows). The graph demonstrates that aPTT reagents vary in their response to coagulation factors. Reagent 2 has a lower sensitivity than reagent 1 for factor VIII and may be less useful in detecting mild hemophilia A. PT reagents (and instrument combinations) also vary in their responses to extrinsic and common pathway factors and are assessed in a similar manner.
Activated Partial Thromboplastin Time (aPTT)
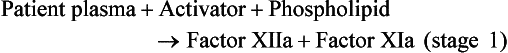
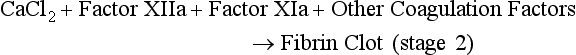
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Hemostasis