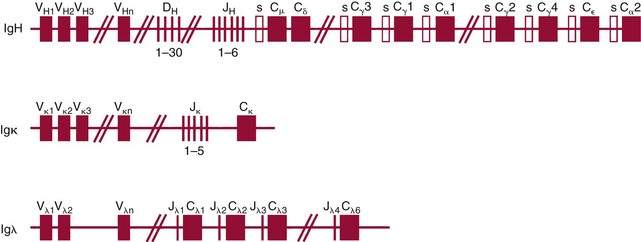
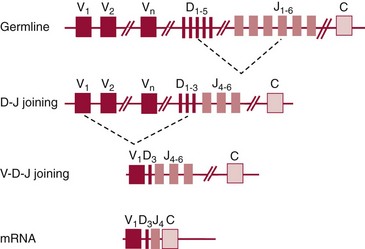
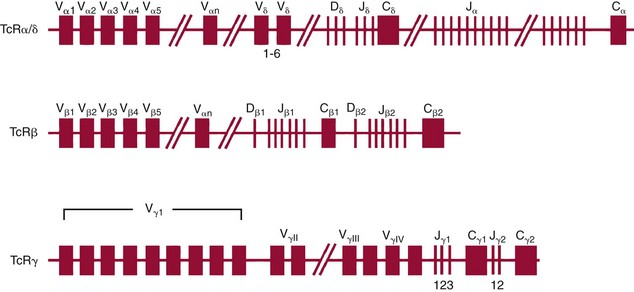
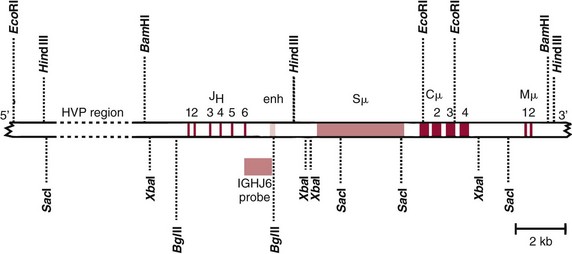
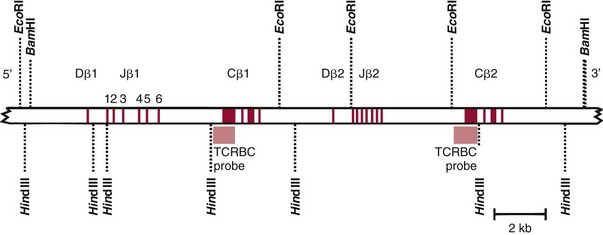
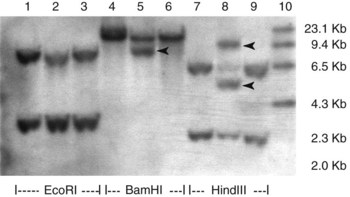
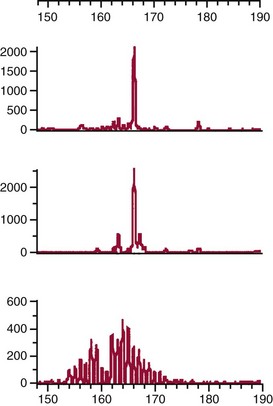
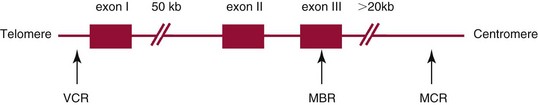
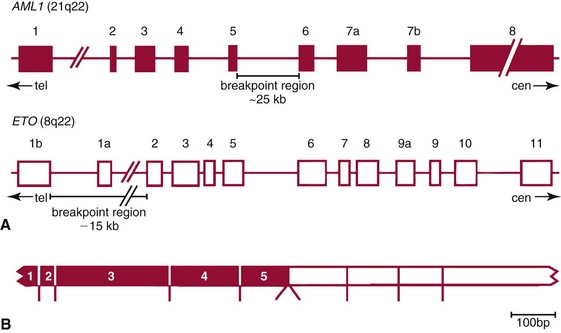
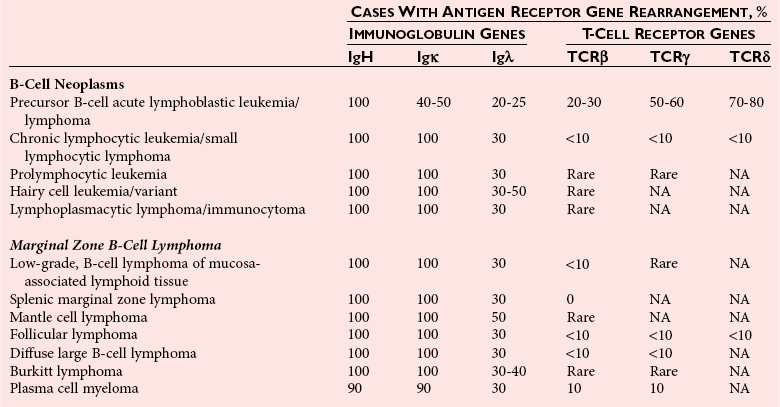
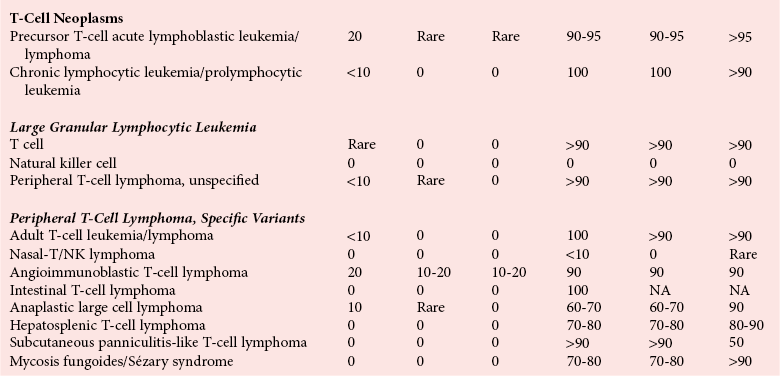
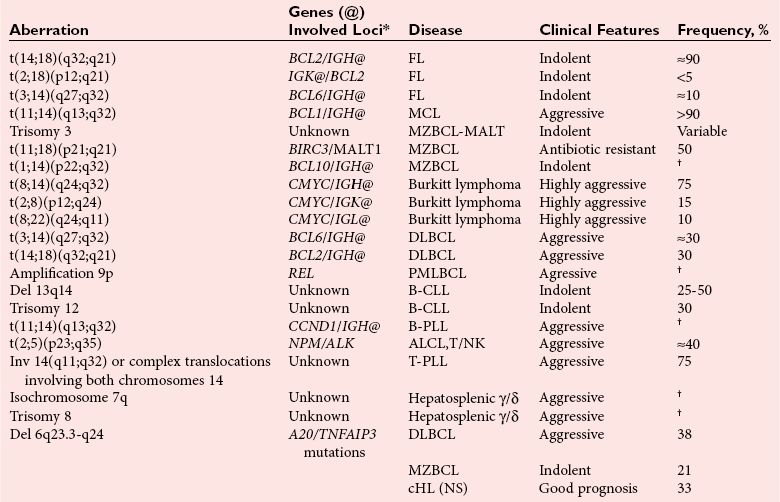
Chapter 44 The histologic features that distinguish benign reactive lymphocytic proliferations from malignant populations can be subtle and somewhat subjective. The introduction of molecular biological techniques into diagnostic pathology has refined lymphoma diagnostics and classification. B and T lymphocytes exhibit the unique property of rearrangement of the immunoglobulin (Ig) and T-cell receptor (TCR) genes, respectively. Immunoglobulin and T-cell receptor genes encode the amino acid sequences of polypeptides that constitute the corresponding antigen receptor. Antigen receptor molecules mediate antigen recognition and are responsible for the specificity of the normal immune response. B lymphocytes are the mediators of humoral response and produce immunoglobulins, and T lymphocytes mediate the cellular response. Each reactive lymphocyte carries a unique antigen receptor gene sequence; therefore, identification of clonal populations arising from a parent malignant lymphocyte becomes feasible because the malignant population is characteristically monoclonal. Tests for monoclonality were originally developed using the Southern blotting hybridization (SBH) method, which remains the gold standard for specificity for detection of clonality throughout a broad range of lymphoid malignancies (Table 44-1). More recently, SBH has been complemented by polymerase chain reaction (PCR)-based strategies. TABLE 44-1 Detection Frequency of Antigen Receptor Gene Rearrangements by Southern Blot Analysis Ig, Immunoglobulin; NA, not applicable; NK, natural killer cell; TCR, T-cell receptor. The organization of Ig genes in the germline is fundamentally similar in all species studied. Genes encoding the two light chains, (κ and λ), and the single locus containing the various heavy chain genes are located on different chromosomes. To generate the large number of antibody molecules needed, B cells have evolved a unique strategy that involves recombination of germline-encoded immunoglobulin gene segments and a somatic hypermutation mechanism that further diversifies the reactivities of the rearranged antigen receptor molecules.140 The immunoglobulin heavy and light chain gene loci—IgH, Igκ, and Igλ—are located at chromosomes 14q32, 2p12, and 22q11, respectively. In the germline configuration, all antigen receptor genes are composed of discontinuous DNA segments that are referred to as variable (V), diversity (D), joining (J), and constant (C) regions. Although both Ig heavy and light chain genes contain V, J, and C regions, only the IgH genes contain D regions (Figure 44-1). The IgH gene contains approximately 87 VH, 30 DH, and 6 JH segments. The IgH VH segment genes can be categorized into seven families, based on relatedness of the DNA sequences. The ability of Ig genes to undergo recombination in a manner analogous to shuffling of cards affords the capacity to generate an enormous primary repertoire of 109 Ig molecules (Figure 44-2). The human Ig C region contains 11 C region segments, which define nine functional immunoglobulin classes and subclasses (IgM, IgD, IgG1, IgG2, IgG3, IgG4, IgA1, IgA2, and IgE). The C region genes are initially uninvolved in the rearrangement process, but are subsequently juxtaposed to the VDJ complex during RNA splicing. Incorporation of C regions into the rearranged VDJ recombination permits class switching. Thus, the specific V(D)J rearrangement can be expressed as IgG, IgA, or other Ig classes. Following the formation of a productive IgH rearrangement, the light chain loci undergo a similar recombination process involving the joining of V to J segments, because no D segments are present in the light chain loci.28 A similar process as described for the Ig genes occurs in the T-cell receptor (TCR) genes, with the notable exception of the absence of a somatic hypermutation mechanism in T-cell receptor gene rearrangements. The developmental hierarchy of T-cell receptor genes is such that the TCR-δ gene is the first to rearrange, followed by the TCRγ, TCRβ, and then the TCRα genes.26 The TCR-δ gene is located on chromosome 14q11 and contains six or fewer Vδ segments, only two or three Dδ segments, and three Jδ segments (Figure 44-3). It is located within the TCR-α gene, immediately 3′ to the last TCR-Vα gene and 5′ to the TCR-Jα segments. This location is important in that productive rearrangements of the TCRδ gene lead to expression of the γ/δ TCR. In the event that the TCRδ rearrangement is nonproductive, Vα to Jα rearrangements obligatorily require that the TCRδ be deleted. The TCRα gene is located on band 14q11 and flanks the TCRδ gene on either side; it contains several Vα segments, an extensive J region composed of up to 50 Jα genes, and one Cα locus. By virtue of the architecture of the TCRα and δ loci, rearrangement of a Vα segment to a Jα segment typically leads to deletion of the intervening TCRδ loci. T cells undergoing such rearrangements may go on to express the TCR-α/β protein. The TCRβ gene is located on band 7q34 and contains 75 to 100 Vβ segments, flanked at the 3′ end by two Dβ, Jβ, and Cβ regions. Each region consists of one Dβ segment, with six or seven Jβ segments and one Cβ segment. Rearrangements may occur within or between segments (e.g., Vβ1 to Jβ1, Vβ2 to Jβ1). Extensive sequence homology is noted between the Cβ1 and Cβ2 segments. This phenomenon is exploited in the utilization of a consensus Cβ probe in the Southern blotting–based clonality assessment of TCR genes. The TCRγ gene is located on band 7q15 and contains about 11 Vγ segments and two Jγ genes.24 The simple structure of the architecture of this gene favors its utilization as a target for TCR receptor PCR, where consensus primers are designed to encompass the relatively few Vγ and the Jγ genes. In this procedure, high-quality total cellular DNA extracted from a fresh or fresh-frozen specimen is subjected to digestion using different bacterial restriction endonucleases, which produce DNA fragments of different sizes encompassing the Ig or TCR gene region segments to be interrogated (Figures 44-4 and 44-5). Enzyme-restricted DNA from each enzyme is subjected to gel electrophoresis and is transferred by blotting and immobilized on a membrane. A labeled probe complementary to the Ig J region segment is hybridized to the membrane. Clonal rearrangements are recognized by the identification of one or two novel rearranged bands that are distinct from the germline pattern obtained with a benign sample or placental DNA. One or two novel rearranged (nongermline) bands may be identifiable, depending on whether a clonal monoallelic or biallelic rearrangement is present (Figure 44-6). The diagnostic hallmark of a clonal population is the presence a nongermline novel rearranged band (see Figure 44-6). However, it is important to recognize that under certain circumstances, nongermline bands do not indicate monoclonality. For example, single-nucleotide polymorphisms (SNPs) may create or abolish restriction enzyme recognition sites, thus yielding fragments with novel restriction banding patterns. In this scenario, utilization of a different enzyme would yield a germline configuration. The potential of these SNPs to confound the interpretation of Southern blot studies justifies the utilization of up to three or more enzymes for the unequivocal assignment of monoclonality. A second situation in which a nongermline band may occur and yet not indicate monoclonality is partial digestion. This is typically evident in the control sample (placental DNA) and invalidates the significance of any such band in the test sample. Clonal rearrangements of the Ig or TCR genes have been used to ascertain the lineage of hematologic neoplasms that do not express B- or T-cell specific markers. Intuitively, clonal rearrangements of Ig genes would indicate B-cell processes, and clonal rearrangements of TCR genes would indicate T-cell processes. However, some malignant lymphomas and leukemias may demonstrate both Ig and TCR rearrangements. In particular, acute leukemias and lymphoblastic lymphomas may demonstrate both Ig and TCR gene rearrangements. Further, up to 20% of acute myeloid leukemias may demonstrate rearrangements of Ig or TCR genes.20,59,73 Hence, lineage is best assigned using a combination of immunophenotypical and molecular studies. Application of PCR for the identification of clonality entails the utilization of consensus V region and J region primers and in vitro amplification of DNA across the V(D)J junction, followed by gel electrophoresis or other methods to determine the size distribution of PCR-generated fragments. In the germline configuration, V and J region segments are located several kilobases apart and are not amplifiable. However, the V(D)J recombination brings the VDJ segments into close proximity, thereby permitting amplification of products that, based on primer location, are usually 80 to 350 bases in length. The V region primers typically are designed to be complementary to the conserved framework (I, II, or III), and the J region primer is typically complementary to consensus sequences present across all six J regions in the immunoglobulin heavy chain gene. Similar strategies may be used for light chain genes as well, but the yield is lower; hence, IgH-PCR is favored. Utilization of framework III consensus primers offers the highest yield but can be complemented by increased diagnostic sensitivity afforded by the inclusion of framework II and/or framework I primer-mediated IgH-PCR for clonality. With regard to T-cell assays, the TCRγ-PCR is the most informative. In these assays, monoclonal populations yield one or two dominant bands (Figure 44-7), while polyclonal cells, each of which carries a unique rearrangement with slightly different band sizes, demonstrate a ladder or smear pattern on gel electrophoresis.81,130 Thus the specificity of each rearrangement identified by PCR is determined by the junctional sequence; hence the marker of clonality in PCR differs from that in SBH analysis, wherein the marker is the configuration of rearranged antigen receptor genes. Following PCR, the amplification products are analyzed by agarose or polyacrylamide gel electrophoresis and are visualized by ethidium bromide staining.38,138 Agarose gels, although less expensive, do not provide resolution of polyacrylamide, which can be configured to reliably discriminate bands differing in size by as few as three bases. This is often very useful in the assessment of PCR products for antigen receptor gene rearrangement assays. Further resolution may be achieved by evaluating sequence-specific and melting characteristics of double-stranded DNA using denaturation gradient gel electrophoresis or temperature gradient gel electrophoresis.55 More recently, capillary gel electrophoresis has become a preferred method for analysis of antigen receptor gene rearrangement. A successful study would exhibit PCR products in the expected size range, appropriate results in positive and negative controls, and absence of bands in the template-free (H2O) control. PCR-based clonality assays are less sensitive in detecting all possible clonal rearrangements when compared with SBH analysis, which approaches 100% if sufficient restriction enzymes and a variety of probes are used. Depending on the assay and the lymphoid neoplasm tested, PCR-based clonality studies may show false-negative rates from 10 to 40% when compared with SBH. False negatives occur for several reasons. First, in contrast to SBH, PCR does not detect partial DJ rearrangements, wherein V and J region genes are not approximated to one another and are too distant for conventional PCR to generate an amplification product. Second, the consensus primers are incapable of hybridizing to all of the different V regions, any one of which could be potentially present in any single neoplasm. Of particular importance to the Ig genes, somatic mutations occurring within the Ig V region genes result in decreased ability for the consensus V region primers to anneal optimally to the V region genes in the template DNA. Thus lymphoid neoplasias with a high somatic hypermutation rate, such as follicular lymphoma, yield lower positive rates by PCR-based Ig clonality tests.125,126 Most of the difficulties described previously can be overcome by the utilization of multiple primer sets. In the event of a negative PCR result, SBH analysis may be performed if sufficient high-quality genomic DNA is available. The main drawback for PCR is the propensity for contamination. Various strategies for contamination control such as maintaining separate DNA extraction and amplification rooms, utilization of laminar flow hoods, and incorporation of uracil DNA glycosylase into PCR should be employed as a rule to prevent contamination problems. The ability to detect rare DNA species carrying a specific molecular aberration (sensitivity) of PCR is dependent on the particular application. For antigen receptor gene rearrangement studies, PCR can detect a monoclonal population in a background of 102 to 103 polyclonal cells of the same phenotype (B or T cells). By comparison, PCR can detect one cell harboring a chromosomal translocation within a background of 105 to 106cells negative for the translocation.92 Antigen receptor gene rearrangements, which are integral to normal B- and T-cell development, and the elaboration of an appropriate immune response are fraught with susceptibility for abnormal translocation of foreign genes, deregulation of which contributes to the development of lymphoid neoplasia. A translocation typically involves the transfer of chromosomal material from one chromosome to a different chromosome. Indeed, nonrandom chromosomal translocations are frequently observed in hematologic neoplasia, and in many cases are characteristic of specific types of tumors. Translocations typically occur in one of two structural configurations, each with distinctive genetic consequences. In the first scenario, the translocation results in the juxtaposition of the intact coding sequence of an oncogene in close proximity with an antigen receptor gene, leading to dysregulated expression and increased production of the structurally normal oncoprotein. This mechanism is most frequently seen in malignant lymphomas, as exemplified by the t(14;18)(q32;q21) characteristic of follicular lymphoma. This translocation juxtaposes the BCL2 gene on 18q21 adjacent to the IgH gene on 14q32, leading to overexpression of BCL2 mRNA and protein.142 In the second scenario, chromosomal translocation leads to an in-frame juxtaposition of two different genes, with resultant creation of a novel chimeric gene. This mechanism is most commonly observed in the leukemias, and less commonly in malignant lymphomas. The t(2;5)(2p23;q35) seen in anaplastic large cell lymphoma is one example wherein an abnormal fusion gene (NPM-ALK) is formed and is central to pathogenesis.96 As another example, the t(11;18)(q21;q21) seen in a subset of extranodal marginal zone B-cell lymphomas of mucosa-associated lymphoid tissue leads to formation of an abnormal API-MALT1 chimeric gene fusion.29 Reverse transcription PCR is a well-suited strategy for the diagnostic detection of such chimeric fusions, because the fusion product is often within the size range of amplifiability of conventional PCR. Many of the translocation breakpoints are widely dispersed and are not clustered. If DNA were to be considered as the template for such PCR, it would best be performed as a long-distance (LD) PCR protocol. However, LD PCR protocols are not applicable to many clinical sample types. A list of common chromosomal translocations, participating genes, and associated lymphomas is provided in Table 44-2. TABLE 44-2 Common Recurrent Genetic Alterations in Human Malignant Lymphoma *Note that IGK and IGL are symbols for genes Igκ and Igλ, respectively. †An insufficient number of patients were studied to permit accurate assessment of percentage of tumors with the cytogenetic aberration indicated. The translocation t(14;18)(q32;21), characteristic of follicular lymphoma, exemplifies the utility of PCR in the detection of a chromosomal translocation characteristic of a specific form of non-Hodgkin lymphoma. The t(14;18) is a balanced reciprocal translocation involving the BCL2 gene (18q21) and the IgH locus (14q32) (Figure 44-8). The BCL2 gene is composed of three exons and two introns.128 At up to four different regions on 18q21, breakpoints are frequently found. The 18q21 breakpoints in most translocations are clustered in the major breakpoint cluster region (MBR). The MBR is a 150 bp sequence located within the 3′ untranslated region of exon 3 of the BCL2 gene, and harbors approximately 60% of the breakpoints found in follicular lymphoma.141,152 Another 20% of follicular lymphomas harbor t(14;18) aberrations in which the breakpoint is located at the minor cluster region, which is located 30 kb further 3′ to the BCL2 gene. An additional cluster of breakpoints occur within the variant cluster region located 5′ upstream to the BCL2 gene, accounting for approximately 5% of the breakpoints found in follicular lymphoma (see Figure 44-8).112 More recently, an intermediate cluster region (ICR) has been described with additional 18q21 breakpoints associated with follicular lymphoma.1 Although initial studies comparing conventional cytogenetics, Southern blot, and PCR-based methods for detection of the t(14;18) (not including the ICR) showed that 86% of cases were identified by Southern blotting and 75% by PCR,66 the inclusion of PCR assays targeting the ICR improves the PCR detection rate for the t(14;18).1 Figure 44-9 shows fluorescence in situ hybridization (FISH) analysis demonstrating the t(14;18) in a sample that tested negative by PCR. An important caveat to note is that the t(14;18) has been detected in benign tonsils and reactive lymph nodes,4,87 and in the peripheral blood of normal blood donors. The t(11;14)(q13;32) abnormality is characteristic of mantle cell lymphoma,85,151 but is rare in other types of malignant lymphoma. This translocation juxtaposes the CCND1 locus on 11q13 with the immunoglobulin heavy chain gene enhancer locus at 14q32.143 This leads to deregulation of the cyclin D1 gene with a quantitative increase in the production of normal cyclin D1 transcripts. Structurally, multiple breakpoints are found on chromosome 11. The most frequently involved location on 11q13 is the major translocation cluster (MTC) region, located 110 kb 5′ to the CCND1 locus.143 Other breakpoints occurring within minor translocation cluster regions have been described 3′ to the major translocation cluster but 5′ to the CCND1 locus (Figure 44-10).111 The CCND1 gene is composed of five exons covering a genomic distance of approximately 15 kb.97 The gene encodes cyclin D1, which regulates cellular transition from G1 to S phase. Despite the diversity of breakpoints flanking the CCND1 gene, the t(11;14) does not lead to disruption of the coding region of the CCND1 gene. Consequently, the expression of cyclin D1 is qualitatively normal but quantitatively increased. One of the practical consequences of the multiple breakpoints in mantle cell lymphoma is that approximately 70% of cases of mantle cell lymphoma can be demonstrated to harbor the t(11;14) by Southern blotting analysis using multiple probes for detection of the various breakpoints.155 By comparison, most published PCR protocols detect only approximately 40% of the t(11;14) anomalies in mantle cell lymphoma, and most of these are clustered in the MTC.155 This is in marked contrast to DNA fiber FISH, which reportedly may detect up to 95% of the t(11;14) aberrations, making FISH the most sensitive method for detection of this abnormality.145 Human leukemias are characterized by recurrent genetic abnormalities that can be utilized as genetic markers of the specific leukemia. The genetic abnormalities are most often recurrent chromosomal translocations that result in chimeric fusions with dysregulated cellular functions, leading to differentiation block or enhancement of proliferation. The chronic leukemias of myeloid-derived cells are characterized by constitutive activation of tyrosine kinases, which primarily confer an excessive proliferative signal and an increased survival advantage. An example of this is the t(9;22), which results in the BCR-ABL chimeric fusion characteristic of chronic myelogenous leukemia (CML). Inhibition of activated tyrosine kinases is a bonafide therapeutic option, as in the case of imatinib mesylate (Gleevec) in CML. By comparison, the chromosomal translocations in acute myeloid leukemias typically result in loss-of-function aberrations within transcription factors that lead to maturational arrest or differentiation blocks at early stages of hematopoietic development. The preponderance of evidence suggests that genetic aberrations leading to maturational arrest in the acute myeloid leukemias are by themselves insufficient to cause leukemia. The concomitant occurrence of cooperating mutations in the RAS and tyrosine kinase genes FLT3 and C-KIT permits progression to a frank leukemia. In either case, the presence of genetic aberrations provides a target for disease detection and monitoring, and for potential gene-specific therapy. A comprehensive list of translocations and fusions thereby arising in acute leukemias is provided in Table 44-3. The most frequently employed technique for the detection of chimeric transcripts in the molecular laboratory is reverse transcriptase–polymerase chain reaction (RT-PCR). Although it is recognized that the definitive method for detection of point mutations is sequencing, a variety of techniques may be used to screen for the presence of point mutations (e.g., single-strand conformation polymorphism analysis,102,134 denaturation gradient gel electrophoresis,12 fluorescence real-time PCR-based melting curve analysis).39 The translocations that occur in the acute myeloid leukemias target transcription factors or transcriptional coactivators. The transcription factors are involved in the differentiation of hematopoietic cells, and disruption of their normal function by the translocation leads to developmental arrest and maturational block at immature stages of differentiation. Targeted transcription factors include the retinoic acid receptor alpha (RARα) in acute promyelocytic leukemia (APL) and several members of the core binding factor (CBF) complex. Core binding factor is a transcription factor composed of heterodimers including a DNA-binding component known as AML1 (RUNX1/CBFA2/PEBP2A) and a subunit CBFβ that activates the transcription of AML1. CBF is targeted in the t(8;21)43 aberration, which results in the AML/ETO fusion characteristic of acute myelogenous leukemia (AML) with differentiation (AML FAB-M2). CBF is similarly targeted in the inv(16), which leads to generation of a fusion gene (CBFβ/SMMHC) comprised of CBFβ and the smooth muscle myosin heavy chain gene (SMMHC) found in a subset of AML4Eo.88 The multilineage importance of CBF is manifest in a subset of precursor B-cell acute lymphoblastic leukemias, where it is targeted in the t(12;21), which leads to the TEL/AML1 fusion.54 All chimeric proteins that result from chimeric fusions are dominant negative inhibitors of CBF-mediated transcription.49,62 The transcriptional repression of CBF target genes caused by CBF-chimeric proteins partially involves recruitment of the histone deacetylase complex. By comparison, APL is characterized by translocations involving the RARα gene (see Table 44-3). These translocations result in a block of myeloid differentiation at the promyelocytic stage. It has been shown that maturational arrest is due in part to recruitment of the nuclear corepressor–histone deacetylase complex. The ability of all-trans retinoic acid (ATRA) to bind to PML/RARα, resulting in release of the nuclear corepressor complex, explains in part the ability of ATRA to relieve the maturational block in promyelocytes central to the development of APL. The t(8;21)(q22;q22) was originally described in a case of acute leukemia by Janet Rowley in 1973.113 The translocation is found in approximately 7% of cases of de novo AML and is more common in young individuals. The translocation is found in 20 to 40% of AMLs of the FAB-M2 subtype.3 It has been reported infrequently in AML FAB-M1, AML FAB-M4, and rare cases of therapy-associated AML. The translocation leads to fusion of the AML1 gene (acute myeloid leukemia 1 gene), also known as CBFA2 (core binding factor subunit A2) or PEBP2α (polyoma enhancer binding protein 2 subunit α), to the ETO gene (8;21), also known as MTG8 (myeloid translocation gene on chromosome 8). The t(8;21) portends a good prognosis in cases of de novo AML, with a favorable response to treatment with cytosine arabinoside.98 The AML1-ETO chimeric fusion is detectable by RT-PCR in the vast majority of t(8;21)-positive AMLs, thus making RT-PCR a good choice for detection of the t(8;21) in clinical samples. The AML1-ETO fusions join exon 5 of AML1 to exon 2 of ETO (Figure 44-11), and the fusion transcript can be detected in complex translocations in a significant proportion of cases that are t(8;21) negative by conventional cytogenetics.3,94 Specifically, RT-PCR has been reported to detect AML1–ETO fusions in 8 to 12% of AML.3,79,94 It is interesting to note that the AML1–ETO fusion transcript may be detected using sensitive end point PCR assays several years after chemotherapy or bone marrow transplant (BMT), thus limiting its predictive value for relapse in affected patients.70 Quantitative studies using recently developed real-time PCR protocols (Figure 44-12) provide a better indication of increasing transcript copies and hence disease recurrence.
Hematopoietic Malignancies
Antigen Receptor Rearrangements for Determination of Clonality
Molecular Genetic Basis for Immunoglobulin Gene Rearrangements
Molecular Genetic Basis for T-Cell Receptor Gene Rearrangements
Southern Blot Hybridization Analysis for Antigen Receptor Gene Rearrangements
Polymerase Chain Reaction Analysis of Antigen Receptor Gene Rearrangements
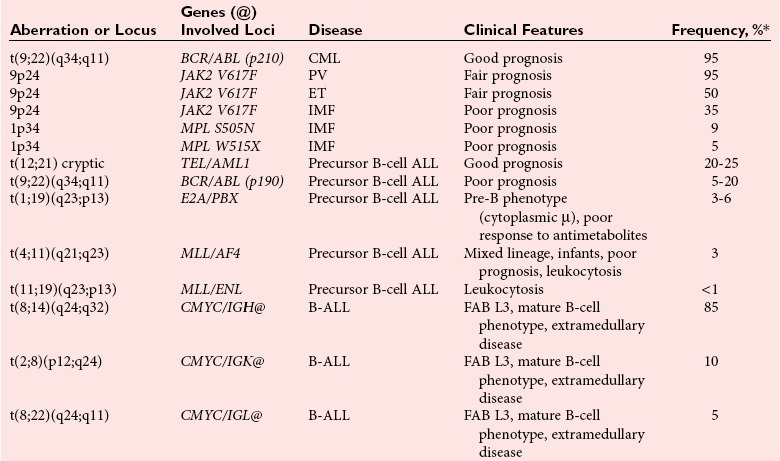
Molecular Genetics of Malignant Lymphomas
Polymerase Chain Reaction Analysis for Detection of Chromosomal Translocations
t(14;18)(q32;21)—BCL2/JH Aberration
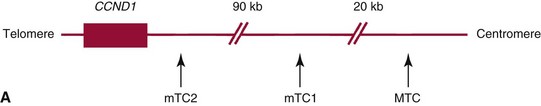
t(11;14)(q13;32)—CCND1/JH Aberration*
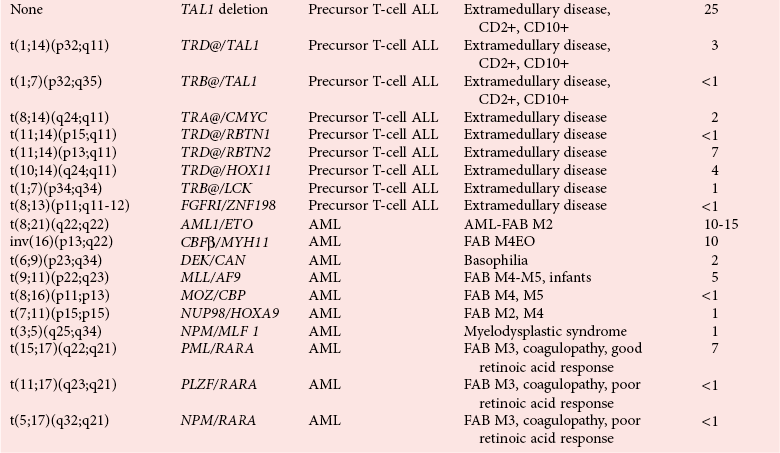
Molecular Genetics of Leukemias
Recurrent Chromosomal Translocations in Acute Myeloid Leukemias
t(8;21)(q22;q22)—AML1-ETO
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Hematopoietic Malignancies