Fig. 14.1
(a, b) H&E, (c) ERG, (d) Inhibin, (e) Carbomic anhydrase and (f) Azocarmine. (a) H&E stain shows a highly vascular neoplasm. The tumor is composed of vascular cells and cells with round nuclei designated as “stromal” cells. (b) Higher power reveals numerous vascular channels (v) and interspersed stromal cells are seen. Note the nuclear pseudoinclusion in a stromal cell (arrowhead). (c): ERG immunohistochemistry. Note the intense nuclear staining in vascular cells; (d, e) Inhibin and carbonic anhydrase immunohistochemistry. Note the intense staining in stromal cells (f) Azocarmine stain highlights vascular channels (a, f, ×50; b–d, and e, ×100).
Histologically, hemangioblastomas are classified into two variants: the more common reticular variant (composed of proliferating vascular elements) and the rare cellular variant (composed of epitheloid clusters of stromal cells), which are associated with greater GFAP positivity, higher proliferation index, and probability of recurrence [39].
Receptors for cellular growth factors including pro-angiogenic factors, such as epidermal growth factor receptor (EGFR), platelet derived growth factor receptor (PDGFR), placental growth factor receptor (PFG-1), and vascular endothelial growth factor receptor (VEGF), are expressed on tumor cells in hemangioblastomas [40]. However, unlike malignant gliomas, the VEGF expression does not correlate with the vascular density as indicated by the expression of CD34-positive endothelial cells. This suggests that pro-angiogenic factors other than VEGF probably contribute to the intense tumor vascularity [41].
Clinical Features
Hemangioblastomas most commonly arise in the CNS especially, but not exclusively, in the posterior fossa. The frequent sites of occurrence in the order of commonality are cerebellum, dorsal part of the spinal cord, brainstem, and retina (Figs. 14.2 and 14.3) [42–44]. The most common site of occurrence of hemangioblastomas in the spinal cord is the thoracic region, followed by cervical and lumbar (48 %, 36 % and 16 %, respectively) [45]. Spinal cord and brainstem hemangioblastomas are frequently associated with tumors at other sites and especially cerebellar hemangioblastomas; in turn, however, cerebellar hemangioblastomas are less frequently associated with tumors at the other sites, suggesting that the spinal cord/brainstem hemangioblastomas are the accompanying manifestation of the latter [46, 47]. Supratentorial (cerebral, sellar/suprasellar, intraventricular) hemangioblastomas are rare [48–50]. It is sometimes difficult to differentiate supratentorial hemangioblastoma from meningioma [38, 51]. Occasionally, hemangioblastomas may arise in extraneural sites such as bone, soft tissue, skin, liver, pancreas, and kidney [52–55].
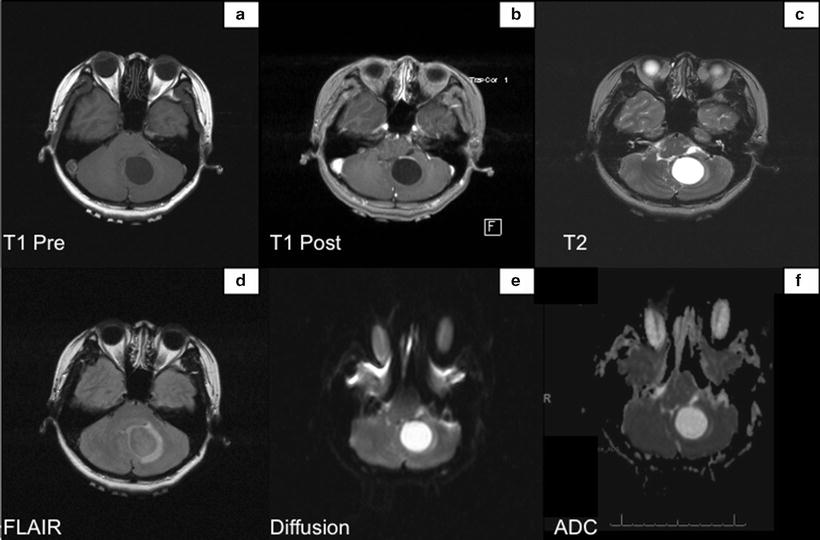
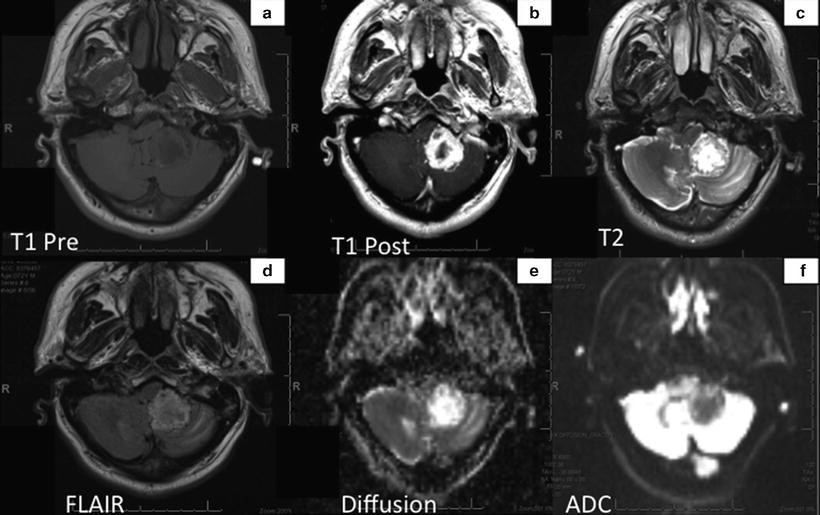
Fig. 14.2
MRI shows the tumor within the inferior medial left cerebellum. Lesion in isointense with the adjacent brain parenchyma on the T1 weighted sequences (Panel a), hyperintense on T2 weighted sequence (Panel c), and avidly enhances gadolinium (Panel b). Diffusion weighted sequence does not demonstrate hyperintense signal within the mass (Panels e and f)
Fig. 14.3
MRI shows the tumor that appears as an irregular thick-walled mass in the region of the left cerebellar tonsil, which enhances intensely with gadolinium (Panel b). Mass in predominantly T1 hypointense (panel a) but contains several areas of T2 hyperintensity indicating hemorrhagic component (Panel c). Diffusion studies show hyperintensity relative to the contralateral white matter (Panels e and f).
One-third of hemangioblastomas are associated with the vHL syndrome. The spectrum of tumors [56] associated with vHL is broad and includes hemangioblastomas, renal cell carcinomas [57, 58], pheochromocytomas [59], extra-adrenal paragangliomas [60, 61], retinal angiomas [62–64], neuroendocrine pancreatic tumors [65–69], papillary cystadenomas of the epididymis [70] and broad uterine ligament [71], as well as endolymphatic sac tumors (ELSTs) of the middle ear [72–74]. vHL-mutated patients with hemangioblastomas are generally younger and present with multiple tumors, while the non-vHL-associated tumors are seen in older patients and are usually solitary.
Based on clinical manifestations, vHL is classified into type 1 and type 2. Type 1 vHL is not associated with pheochromocytoma while type 2 is. Type 2 is further divided into type 2A, 2B, and 2C. vHL-type 2b is associated with high incidence of hemangioblastoma and pheochromocytoma [44, 75–77].
Since patients with vHL syndrome are predisposed to developing multiple hemangioblastomas and require specialized surveillance and treatment, it is imperative to correctly diagnose vHL as early as possible. Genetic testing for vHL in addition to a comprehensive family history should be considered standard practice for all patients with CNS hemangioblastomas, especially those diagnosed under 30 years of age. Clinical screening of vHL-associated tumors consists of complete neuraxis imaging with magnetic resonance imaging (MRI) of the brain and entire spine, MRI of the abdomen, retinoscopy, and measurement of urine catecholamines. Some authors have suggested ophthalmologic screening for family members of vHL disease for early detection of retinal hemangioblastomas [78].
Hemangioblastomas are considered benign tumors, but can cause significant neurological deficits depending on their size and location. Headache, vomiting, cerebellar symptoms, and cranial nerve involvement may be the presenting features. Posterior fossa tumors can also cause cerebrospinal fluid (CSF) flow obstruction leading to hydrocephalus [79, 80]. Patients with spinal cord tumors may present with progressive scoliosis and radicular symptoms until the tumor is large enough to cause weakness. Onset of retinal hemangioblastomas can start prior to 10 years of age until 30 years, after which the risk gradually decreases. It usually presents with unilateral involvement [77]. Hemangioblastomas exhibit a stuttering growth pattern, i.e., there are periods of growth followed by periods of quiescence, which may be as long as 2 years. Indications for treatment relate to the patient’s symptoms and tumor size, location, and rate of progression [81]. It is quite common for spinal cord hemangioblastomas to present with syrinx formation [82]. Occurrence of erythrocytosis with male predominance is common in hemangioblastomas due to production of erythropoietin [83, 84]. Due to their arteriovenous malformation-like vascularization, solid hemangioblastomas present a unique neurosurgical challenge [85].
There have been numerous clinical reports of worsening of vHL-associated hemangioblastomas in pregnancy, leading to progressive neurological deficits and obstructive hydrocephalus [86–90]. However, in the first prospective study comparing the rate of tumor growth in pregnant versus the nonpregnant cohorts with vHL-associated hemangioblastomas, Ye et al. observed that there were no differences in tumor growth rate, peritumoral cyst growth and the need for surgery. However, this was a small study with only 27 patients in the pregnancy cohort and it is possible that patients who chose to become pregnant were already in a better state of health leading to selection bias [91].
Imaging
Hemangioblastomas show post-contrast enhancement on computed tomogram (CT) scans and T1-weighed MRI. Imaging studies show the typical appearance of a cyst with mural nodule in approximately 60 % of cases. The nodular portion shows flow voids in the T1 and T2-weighted sequences. Generally, the cysts are slightly hyperintense compared to CSF in T1-weighted images. Both the nodule and the cyst appear bright on T2 and fluid attenuated inversion recovery (FLAIR) sequences [92].
Treatment
While most neurosurgeons agree that surgical intervention of symptomatic hemangioblastomas is required, controversy arises in dealing with asymptomatic hemangioblastomas, which commonly occur in patients with vHL syndrome. Unlike other benign intracranial tumors that exhibit a slow, progressive growth pattern, hemangioblastomas often have prolonged periods of growth arrest, thus making their natural course difficult to predict [81]. For asymptomatic, radiographically stable tumors, no treatment may be recommended. When asymptomatic tumors show progression on imaging only, the best time for intervention may be difficult to determine [93–96]. Similar to patients with other tumor predisposition syndromes, the optimal clinical management of vHL requires a specialist who oversees and coordinates a multidisciplinary plan of care, including appropriate screening tests.
From a therapeutic perspective, surgical removal remains the treatment of choice for hemangioblastomas and has been successfully employed for cerebellar [97], spinal [98, 99], and brainstem [94, 100] hemangioblastomas. Preoperative cerebral angiography helps surgeons determine the nature of the tumor vascular supply. Following diagnostic imaging, pretreatment with dexamethasone for several days is generally recommended. Intraoperative bleeding increases with tumor size, making en bloc resection of larger tumors difficult. However, modern microsurgical techniques are used to identify feeding vessels and thus help minimize intraoperative bleeding. Dissection should be carried out along the external surface of the tumor in the gliotic brain-tumor interface, to avoid entering the tumor, thus preventing brisk hemorrhage from the hemangioblastoma. The tumor-associated cysts are non-neoplastic and consist of compressed glial tissue, which collapses on its own once the associated tumor is removed. Postoperative complications include temporary worsening of neurological deficits, new neurological deficits, which may or may not resolve during follow-up, cranial postoperative infection, hydrocephalus and aseptic meningitis [101]. A postoperative contrast-enhanced MRI is routinely obtained to verify extent of resection. If no residual is noted, tumor recurrence is rare.
More recently, stereotactic radiosurgery is also being employed with encouraging results especially in spinal hemangioblastomas [102–105]. One advantage of radiosurgery is the ability to treat multiple lesions in a single treatment setting. In a series of 9 patients with 20 spinal hemangioblastomas, 4-year tumor overall and solid tumor control rates with stereotactic radiosurgery were as high as 90 % and 95 %, respectively [106]. In other studies, however, patients with multiple hemangioblastomas associated with vHL syndrome were found to less likely exhibit tumor control after treatment with radiation therapy compared to single sporadic hemangioblastomas [107, 108]. In general, smaller tumor volumes and higher doses of radiation (median 16 Gy) confer a better tumor control [109].
In contrast to surgery and radiation therapy, there is a paucity of data on systemic treatment of hemangioblastomas. Since hemangioblastomas are highly vascular, systemic anti-angiogenic therapies are being investigated as an alternative to surgery, particularly in vHL patients with multiple tumors. Several vHL patients have been treated with semaxanib, a multi-tyrosine kinase inhibitor predominantly active against VEGFR-2. Although disease stabilization outside the CNS was observed in some patients, most of the treatment responses were limited to retinal hemangioblastomas [110]. In a clinical trial for vHL patients with sunitinib, which predominantly targets VEGFR and PDGFR, antitumor activity was seen against renal cell carcinoma, but not hemangioblastomas [111]. EGFR, which is overexpressed and activated in hemangioblastomas, represents an additional attractive target for therapeutic intervention and study in future clinical trials [112]. There have been case reports on the use of anti-angiogenic agents such as bevacizumab [113], pazopanib [114], sunitinib [115], thalidomide [116], and interferon [117] with limited success. However, no prospective clinical trials using these agents have been conducted to date.
Prognosis
Gross total tumor resection was a predictor of prolonged progression-free survival (PFS) in one series [118]. Poor prognostic factors include poor performance status [101], multiple hemangioblastomas, retinal hemangioblastomas, and presence of solid rather than cystic tumors. The risk of recurrence in the future is higher if the age of diagnosis is younger than 40 years with primary sites being the brainstem and spinal cord [119].
Acknowledgment
The authors thank Dr Ajax George, Department of Radiology, NYU Langone Medical Center, for providing MRI images.
References
1.
Surawicz TS, Mccarthy BJ, Kupelian V, Jukich PJ, Bruner JM, Davis FG. Descriptive epidemiology of primary brain and CNS tumors: results from the Central Brain Tumor Registry of the United States, 1990–1994. Neuro Oncol. 1999;1:14–25.PubMedCentralPubMed
2.
Richard S, Beigelman C, Gerber S, van Effenterre R, Gaudric A, Sahel M, Binaghi M, DE Kersaint-Gilly A, Houtteville JP, Brunon JP, et al. Does hemangioblastoma exist outside von Hippel-Lindau disease? Neurochirurgie. 1994;40:145–54.PubMed
3.
Sora S, Ueki K, Saito N, Kawahara N, Shitara N, Kirino T. Incidence of von Hippel-Lindau disease in hemangioblastoma patients: the University of Tokyo Hospital experience from 1954–1998. Acta Neurochir (Wien). 2001;143:893–6.
4.
Sprenger SH, Gijtenbeek JM, Wesseling P, Sciot R, van Calenbergh F, Lammens M, Jeuken JW. Characteristic chromosomal aberrations in sporadic cerebellar hemangioblastomas revealed by comparative genomic hybridization. J Neurooncol. 2001;52:241–7.PubMed
5.
Gijtenbeek JM, Jacobs B, Sprenger SH, Eleveld MJ, van Kessel AG, Kros JM, Sciot R, van Calenbergh F, Wesseling P, Jeuken JW. Analysis of von hippel-lindau mutations with comparative genomic hybridization in sporadic and hereditary hemangioblastomas: possible genetic heterogeneity. J Neurosurg. 2002;97:977–82.PubMed
6.
Prowse AH, Webster AR, Richards FM, Richard S, Olschwang S, Resche F, Affara NA, Maher ER. Somatic inactivation of the VHL gene in Von Hippel-Lindau disease tumors. Am J Hum Genet. 1997;60:765–71.PubMedCentralPubMed
7.
Rickert CH, Hasselblatt M, Jeibmann A, Paulus W. Cellular and reticular variants of hemangioblastoma differ in their cytogenetic profiles. Hum Pathol. 2006;37:1452–7.PubMed
8.
Lemeta S, Pylkkanen L, Sainio M, Niemela M, Saarikoski S, Husgafvel-Pursiainen K, Bohling T. Loss of heterozygosity at 6q is frequent and concurrent with 3p loss in sporadic and familial capillary hemangioblastomas. J Neuropathol Exp Neurol. 2004;63:1072–9.PubMed
9.
Lemeta S, Salmenkivi K, Pylkkanen L, Sainio M, Saarikoski ST, Arola J, Heikkila P, Haglund C, Husgafvel-Pursiainen K, Bohling T. Frequent loss of heterozygosity at 6q in pheochromocytoma. Hum Pathol. 2006;37:749–54.PubMed
10.
Lemeta S, Jarmalaite S, Pylkkanen L, Bohling T, Husgafvel-Pursiainen K. Preferential loss of the nonimprinted allele for the ZAC1 tumor suppressor gene in human capillary hemangioblastoma. J Neuropathol Exp Neurol. 2007;66:860–7.PubMed
11.
Lonergan KM, Iliopoulos O, Ohh M, Kamura T, Conaway RC, Conaway JW, Kaelin Jr WG. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol. 1998;18:732–41.PubMedCentralPubMed
12.
Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol. 2008;9:285–96.PubMedCentralPubMed
13.
Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–7.PubMed
14.
Kaelin Jr WG. The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem Biophys Res Commun. 2005;338:627–38.PubMed
15.
Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A. 2000;97:10430–5.PubMedCentralPubMed
16.
Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol. 2009;19:12–6.PubMed
17.
Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275:25733–41.PubMed
18.
Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19:4298–309.PubMedCentralPubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


