The practicing surgical pathologist is now likely to encounter specimens beyond valve leaflets and pericardiectomy specimens. In centers with active interventional, transplant, and surgical programs, endomyocardial biopsies (EMBs), ventricular core excisions from ventricular assist device (VAD) placement, septal myomectomies, tumor excisions, prosthetic valve and VADs, and native hearts are encountered. In each case, careful attention to specimen handling and preparation is essential, and the specific requirements will be addressed in each category (68).
MYOCARDIAL DISEASES
Over the past 50 years, the terminology, concepts, and classification of myocardial disorders have undergone numerous revisions. The World Health Organization (WHO) originally defined the term cardiomyopathy in 1968 as “diseases of different and often unknown etiology in which the dominant feature is cardiomegaly and heart failure” (69). This functional definition was modified in the 1980 WHO classification to “heart diseases of unknown cause” (70). The three types of primary cardiomyopathy consisted of dilated or congestive, hypertrophic (with or without obstruction), and restrictive types. The 1995 classification reflected new knowledge of disease genetics, etiologies, and pathogenesis and expanded the scope to “diseases of the myocardium associated with cardiac dysfunction” (71). Primary cardiomyopathies and specific heart muscle diseases were enumerated. In 2006, a consensus group of the American Heart Association (AHA) defined cardiomyopathies as “a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are a result of a variety of causes that are frequently genetic” (72). In this etiologic/pathogenetic classification, there are primary types that are genetic, nongenetic, or acquired in nature and secondary types that are usually a component of a systemic disease (Table 29.1). In 2008, the European Society of Cardiology (ESC) proposed a new classification based on the definition of cardiomyopathy as a “myocardial disorder in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular heart disease and congenital heart disease sufficient to cause the observed myocardial abnormality” (73). This clinically oriented classification established familial and nonfamilial categories for each of the major types of cardiomyopathies and eliminated the primary and secondary types (Table 29.1). In 2013, the World Heart Federation endorsed a novel genotype/phenotype approach to the classification of cardiomyopathies that builds on the ESC definition of cardiomyopathy. Five attributes of cardiomyopathy are catalogued in the MOGE(S) scheme: the structural and functional abnormality or phenotype (M), the extent of organ involvement (cardiac vs. extracardiac) (O), the genetic or familial inheritance (G) of the process, the etiology of the genetic defect (E), and the AHA/American College of Cardiology (ACC) stage or New York Heart Association (NYHA) functional classification of heart failure or exercise intolerance (S) (74). Although these recent classifications have their individual merits, we prefer a morphologic approach to cardiomyopathy that highlights the key macroscopic and/or histopathologic findings that can be identified in EMBs or larger specimens such as ventricular apical plugs and explanted hearts. In many cases, histochemical and immunohistochemical stains and molecular studies aid in the classification. In some disorders such as dilated cardiomyopathy (DCM), the histopathologic features are nonspecific, and the role of EMB is to exclude treatable causes of ventricular dysfunction (Table 29.2).
THE NORMAL HEART
The macroscopic, histologic, and ultrastructural features of the normal heart have been described in several textbooks and monographs and will only be briefly reviewed here (75,76). The myocardium is composed of bundles of myocytes separated by fibrous bands. Individual myocytes form a syncytium connected by intercalated disks at their terminal ends and occasionally display side-to-side junctions. On average, myocytes measure 15 to 17 μm in diameter and display central round to oval nuclei measuring up to 8 μm in diameter. Lipofuscin granules can be seen in a perinuclear position. The cytoplasm contains actin and myosin filaments, mitochondria, glycogen granules, and Z-bands. The interstitium is composed of collagen and elastic fibers, a proteoglycan- and glycosaminoglycan-rich matrix, basement membrane proteins such as laminin and fibronectin, and a rich capillary vascular network admixed with scattered fibroblasts, leukocytes, and, on occasion, adipocytes and nerve twigs (77).
THE ENDOMYOCARDIAL BIOPSY
In many centers, the evaluation of cardiomyopathic patients includes a detailed history and physical examination, many of the sophisticated techniques described earlier, coronary angiography, and EMB. The indications for EMB have been previously published, including a recent consensus statement (78–83). Currently, the major indications include the diagnosis of acute transplantation rejection, the classification of myocarditis, the evaluation of recent-onset heart failure in the absence of coronary artery disease, the grading of anthracycline cardiotoxicity, the evaluation of restrictive heart disease such as amyloidosis, and the diagnosis of storage disorders and cardiac tumors. Complications are uncommon and include hematomas and nerve injury and cardiac problems such as arrhythmias, tricuspid valve apparatus damage, and ventricular perforation. In most centers, the right ventricular EMB is used, but there are a few published series reporting the use of the left ventricular approach (84,85).
Proper tissue procurement and handling are essential for optimal diagnostic evaluation. Biopsy specimens should be gently extracted from the bioptome with a needle tip to limit crush artifactual distortion. The clinical indications for the biopsy determine, in large part, the method of tissue handling. For example, for standard light microscopy, the tissue should immediately be placed in a standard fixative such as 10% neutral buffered formalin. To subtype amyloid fibrils in cardiac amyloidosis by immunofluorescence (e.g., AL, AA, or transthyretin), one to two pieces should be submitted in saline or Zeus medium and then snap frozen in a plastic Beem capsule containing an optimal cutting temperature (OCT)–embedding medium. Snap freezing tissue is optimal for preserving tissue for molecular analysis such as nested polymerase chain reaction (PCR) and real-time PCR in the evaluation of viral pathogens. The diagnosis of chronic anthracycline cardiotoxicity requires that all the biopsy pieces (minimum of three to five pieces) should be fixed for transmission electron microscopy (e.g., 2.5% glutaraldehyde with 2% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.2).
For routine diagnostic evaluation, overnight processing and paraffin embedding are sufficient. For emergent cases, a 90-minute “ultra” processing cycle can be used, and slides can be made available within 3 to 4 hours. All of the biopsy pieces should be embedded in the same block. We recommend a minimum of three to six slides prepared at 4- to 5-μm thickness from various depths within the paraffin block. Multiple paraffin ribbons are placed on each slide. We routinely stain with hematoxylin and eosin and use stains such as Masson trichrome to confirm the presence of myocyte damage or fibrosis, Congo red or sulfated Alcian blue stain for amyloid fibrils, and the Prussian blue stain for iron deposition. Immunohistochemical, immunofluorescence, and molecular studies are used for specific indications. Paraffin section immunohistochemistry is used to evaluate for infectious myocarditis (e.g., cytomegalovirus [CMV] or toxoplasmic myocarditis), posttransplantation lymphoproliferative disorders (PTLDs) (e.g., B-cell clonality, Epstein-Barr virus [EBV] latent membrane proteins, anomalous coexpression of B-cell and T-cell antigens), or antibody-mediated rejection (intravascular collections of CD68+ histiocytes and deposition of C4d on the microvasculature). In situ hybridization is helpful to demonstrate the presence of EBV or other viral genome or light chain restriction in PTLD.
A variety of artifacts in EMB specimens can mimic pathologic lesions. The surgical pathologist must be aware of these patterns to avoid a misdiagnosis that could lead to unnecessary therapeutic interventions. These have been reviewed in detail in a recent publication, and only selected topics will be briefly reviewed (75,81). The most common biopsy artifact is the presence of contraction bands in myocytes that are identical to the linear bands observed in acute ischemic necrosis and catecholamine (“pressor”) effect. Another frequent artifact is intussusception or “telescoping” of small arteries that has been confused with luminal occlusion by thrombus and transplantation-related arteriosclerosis. Connective tissue stains such as Masson trichrome or elastic van Gieson highlight the internal elastic membranes of both vessel segments. Intramyocardial accumulations of mature adipose tissue can simulate epicardial tissue, especially if associated with vessels of relatively large caliber, and both can be found in the right ventricular (RV) apical region. Ventricular perforation is identified by the presence of mesothelial cells. Accumulations of fresh platelet/fibrin-rich thrombus may be identified along the endocardial surface of biopsy fragments. Bioptome-induced tissue distortion or crush artifact can occur, and on occasion, it may not be possible to distinguish the cell types (e.g., lymphocytes, endothelial cells, histiocytes, myocytes). In some instances, additional leveled hematoxylin and eosin–stained sections provide less distorted foci in the deeper aspects of the biopsy sample.
The surgical pathology report should be comprehensive to provide the clinician with as much diagnostic information as possible. The number of pieces of myocardium, the appearance of the myocyte nuclei (hypertrophied, pyknotic, attenuated, or atrophic), the presence of cytoplasmic pigments or other accumulations, the pattern of necrosis (focal vs. diffuse), the composition of the interstitium (e.g., cellularity, fibrosis, edema, amyloid deposits), and the presence of endocardial inflammation should be addressed. It is essential that detailed clinical information be provided to allow clinicopathologic correlation.
PRIMARY CARDIOMYOPATHIES
Dilated Cardiomyopathy
Dilated or congestive cardiomyopathy is the most common type of idiopathic or primary cardiomyopathy and is one of the most common indications for heart transplantation (86). In the United States, the estimated prevalence in adults is 1 in 2500; it is much less common in children with an estimated prevalence of 1 to 2 in 100,000 (87,88). It is characterized by systolic dysfunction, leading to diminished stroke volumes, elevated left ventricular (LV) end-systolic and end-diastolic volumes, diminished ejection fraction, increased ventricular chamber dimensions and wall tension, and thinning of LV wall (Table 29.3). In most cases, both ventricles are affected, and at the time of transplantation, four-chamber dilatation is common (Fig. 29.3). By definition, significant coronary artery disease, hypertensive heart disease, and valvular causes of contractile dysfunction are absent. The pathogenesis is elusive; approximately 50% of cases are idiopathic, and the remainder is composed of familial cases (25% to 35%), chronic alcohol and heavy metal toxicity, and peripartum causes (86). The genetic determinants are variable, and to date, more than 50 single gene mutations have been linked to familial DCM. These include mutations in genes coding sarcomeric, cytoskeletal, and nuclear proteins and proteins involved in the regulation of calcium metabolism (48,89). The four mutations that currently account for the majority of cases are titin (TTN), lamin A/C (LMNA), beta-myosin heavy chain (MYHT7), and cardiac troponin (TNNT2). The most common pattern of inheritance is autosomal dominant, but the penetrance is variable; other reported patterns include autosomal recessive and X-linked. Interestingly, variants within a particular gene can produce different patterns of cardiomyopathy; for example, mutations in the gene coding the sarcomeric protein cardiac troponin I (TNNI3) can cause dilated, hypertrophic, or restrictive cardiomyopathies (48). Of the idiopathic causes, postviral and other infectious events and autoimmune mechanisms are suspected pathogenetic mechanisms. The clinical manifestations of DCM reflect the poor systolic pump function of the heart. Congestive heart failure symptoms are common. Complications include supraventricular and ventricular arrhythmias, thromboembolic events, and sudden death.
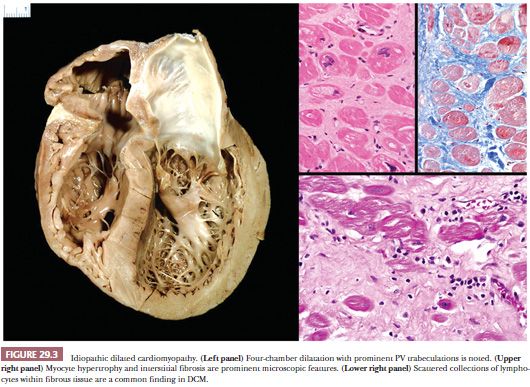
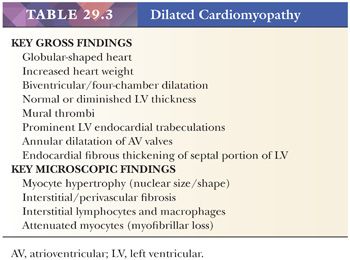
The macroscopic and histopathologic findings result from ventricular remodeling. Increased heart weight, globular shape, chamber dilatation of both ventricles and atria, prominent fine reticulation of LV trabecular musculature, mural thrombi in one or more chambers, endocardial fibrous thickening over the septal portion of the LV, annular dilatation of atrial-ventricular valves, and reduced free wall thickness are common macroscopic findings (Fig. 29.3) (90). These features can be best appreciated by a four-chamber view of the heart or by the short axis (bread loafing) method. The microscopic findings are less dramatic and nonspecific. The myocytes appear attenuated usually with increased vacuolization. Myocyte hypertrophy is characterized by enlarged, irregularly shaped, and hyperchromatic nuclei. Cytoplasmic accumulations of lipofuscin pigment are common. The interstitium is expanded by collagenous fibrosis that ranges from fine to coarse in composition. Perivascular fibrosis is also conspicuous. Scattered collections of mononuclear inflammatory cells are observed in more than 80% of cases, but myocyte damage is absent (91). Nishikawa and colleagues (92) recently reported subtle morphologic differences on EMB between pediatric and adult patients with DCM. At the ultrastructural level, myofibrillar loss; Z-band remnants; and numerous, irregularly shaped mitochondria masses are observed, particularly in the advanced stages of DCM (93).
Several disorders can mimic DCM both clinically and pathologically. The primary role of EMB is to exclude those mimics with specific diagnostic features and therapies such as lymphocytic myocarditis, sarcoidosis, hemochromatosis, anthracycline cardiotoxicity, and so on. A detailed clinical history is required to exclude acquired causes such as collagen vascular disorders, endocrinopathies, nutritional deficiencies, recreational abuse of drugs such as methamphetamines, and tachycardiomyopathy (73). In our experience, the majority of cases of peripartum/postpartum cardiomyopathy that have an EMB as part of the workup show features of DCM. We avoid the term chronic myocarditis for cases with scattered lymphocytes embedded in fibrous tissue because we think that these cases fall within the spectrum of DCM. The number of truly idiopathic cases is likely to continue to diminish with the application of newer and more sophisticated molecular studies.
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is the most common genetic disorder of the heart and affects 1 in 500 adults (48,94). HCM is characterized by myocyte hypertrophy in the absence of abnormal loading disorders such as hypertension and stenotic valvular heart disease or systemic or infiltrative disorders such as amyloidosis or hemochromatosis. It has an autosomal dominant pattern of inheritance with variable phenotypic expression depending in large part on age (95). Patients present across a wide age spectrum, including infants, but most manifest symptoms during adolescence. Symptoms include exertional dyspnea and chest pain, palpitations, light-headedness, syncope, and sudden death; HCM is a common cause of sudden death in young athletes. In some cases, patients are asymptomatic.
In its classic form, HCM is caused by a single gene mutation. In 1990, Geisterfer-Lowrance et al. (96) identified a missense mutation in the beta-myosin heavy chain in a family with HCM. Ten additional mutations that encode sarcomeric proteins have been identified; beta-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC3) are the most frequent point mutations and account for 70% of sarcomeric HCM. Several metabolic and storage diseases, such as glycogen storage disease, carnitine deficiency, Anderson-Fabry disease, and Danon disease; mitochondrial cytopathies such as Senger syndrome and Friedreich ataxia; and syndromic forms of HCM such as Noonan and LEOPARD syndromes produce cardiomyopathies that phenotypically resemble the sarcomeric forms (Table 29.4) (48,74,97).
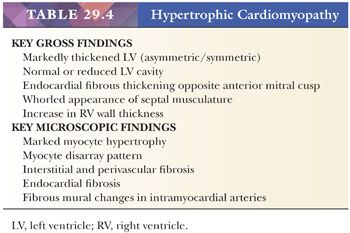
The original pathologic description of HCM by Teare in 1958 (98) provides many of the key morphologic criteria used today. The macroscopic findings are optimally presented by the short axis method of sectioning or in a long axis cut through the LV outflow; the lines-of-flow approach is best avoided (99). Transverse slices of the myocardium demonstrate the markedly thickened LV, usually with disproportionate enlargement of the septum and normal or reduced LV chamber size. The ratio of septal thickness to LV free wall thickness of more than 1.3 is used by some pathologists to confirm asymmetric hypertrophy (100). In many cases, the musculature of the septum displays a whorled arrangement of the muscle bundles (Fig. 29.4). The RV is also thickened in the majority of cases, particularly in the anterior wall of the RV outflow tract. Most cases of HCM display asymmetric thickening of the interventricular septum, but concentric or symmetric hypertrophy is also reported. For this reason, the diagnosis of HCM requires that hypertensive heart disease and amyloidosis be excluded by clinical and morphologic means, respectively. In patients with outflow tract obstruction, the septum may have a transverse fibrous plaque on the endocardium at the site of repetitive contact with the anterior leaflet of the mitral valve. This repetitive mechanical injury results in thickening of the leaflet. In the late stages of HCM, protracted heart failure may lead to LV dilatation and obscure some of these findings. The disrupted muscular arrangement seen grossly reflects the microscopic disorganization and fibrosis within the more central portions of the septum, particularly toward the base of the heart. Myocytes showing marked hypertrophy with pleomorphic and hyperchromatic nuclear forms alternate with haphazard bands of dense collagenous tissue to create the characteristic interwoven myocyte disarray pattern (Fig. 29.4). The loss of orderly myocyte bundles is often prominent at scanning magnification. The trichrome stain highlights the interstitial fibrosis and the subendocardial plaques. The small intramyocardial arteries show disorganized smooth muscle proliferation with reduced lumens and intramural and perivascular fibrosis (101). Foci of coarse interstitial fibrosis can be found within the central portions of the septum.
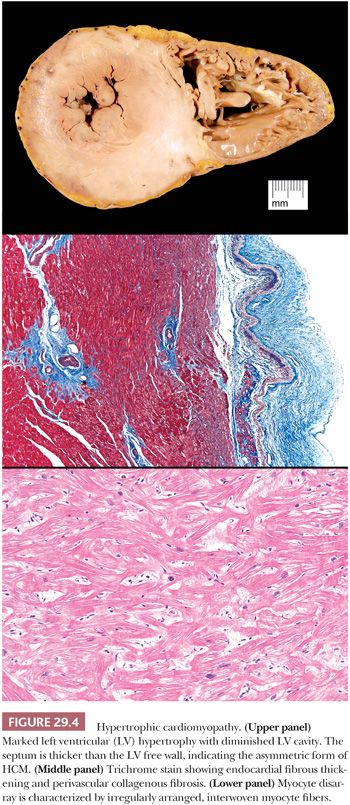
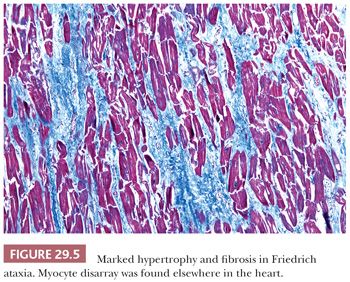
Several points must be emphasized about the differential diagnosis of HCM. First, myocyte disarray is observed in cases of HCM as a result of mutations of genes coding for sarcomeric proteins and in the nonsarcomeric forms such as Friedreich ataxia and Noonan syndrome (Fig. 29.5). On occasion, these changes can also be seen in hypertensive heart disease and in some congenital heart diseases, but they are usually patchy (102). Hypertensive heart disease and amyloidosis can mimic some of the macroscopic features of HCM, and careful attention to clinical history and the microscopic findings is essential. Routine amyloid staining of myomectomy specimens of patients more than 65 years old is advocated by many investigators (103). RV EMB is not helpful in establishing the diagnosis of HCM because myocyte disarray is a normal finding in the RV apex and in trabecular muscles. It is, however, useful to exclude other causes such as amyloidosis and hemochromatosis. Distinction of HCM from “athlete’s heart” is important and at times difficult. Sustained isometric exercise produces a physiologic form of hypertrophy with increased LV muscle mass, LV wall thickening, and normal or reduced chamber dimensions. Importantly, the septal-to-posterior free wall ratio is not greater than 1.3 and the wall thickness does not exceed 1.6 cm in athlete’s heart (104,105).
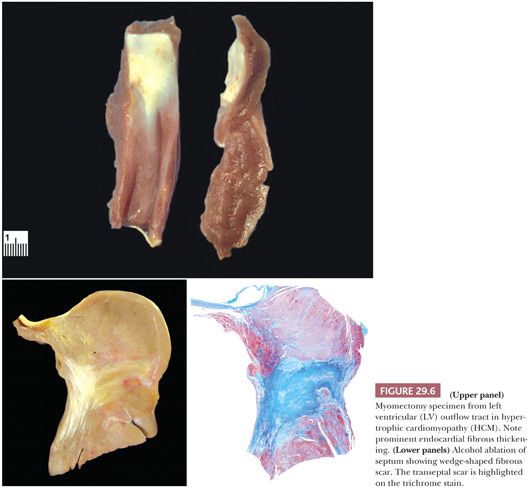
Currently, the medical management of HCM is directed at improving myocardial relaxation and increasing diastolic filling. Ventricular septal myotomy-myomectomy removes the portion of the septum beneath the aortic valve and is indicated in patients who fail medical therapy or have severe outflow obstruction (Fig. 29.6) (106). Tazelaar and Billingham (107) found disarray in 58% of myomectomy specimens along with the interstitial and vascular changes described earlier. In a recent series of 204 cases from the Mayo Clinic, the microscopic findings included myocyte hypertrophy (100%), endocardial (96%) and myocardial fibrosis (93%), myocyte disarray (79%), endocardial inflammation (48%), arterial thickening (46%), dilated venules (28%), and arterial dysplasia (16%) (108). In our experience, myocyte disarray is not found in small or superficial myomectomy specimens because it is generally located in the mid region of the septum. Another therapeutic intervention is transcoronary septal ablation with alcohol (109). The septal perforator branch(es) of the left anterior descending artery is infused with 1 to 3 cc of pure alcohol, causing localized infarction and eventually retraction and scarring of the basal portion of the septum (Fig. 29.6). There are very few studies comparing the two techniques, and a controlled randomized study has not yet been reported (110–113).
Restrictive Cardiomyopathy
The disorders comprising the primary restrictive cardiomyopathies are a heterogeneous group characterized by diastolic dysfunction as a result of thickened or “stiff” ventricles. Secondary causes are more common and include several infiltrative disorders, storage diseases, and postradiation disorder. In the setting of restrictive hemodynamics, ventricular compliance is diminished, causing elevated ventricular filling pressure but preservation of systolic function. Other causes of diastolic dysfunction, such as constrictive pericarditis, need to be excluded by clinical and imaging modalities like echocardiography. The EMB is useful for excluding many of the secondary types.
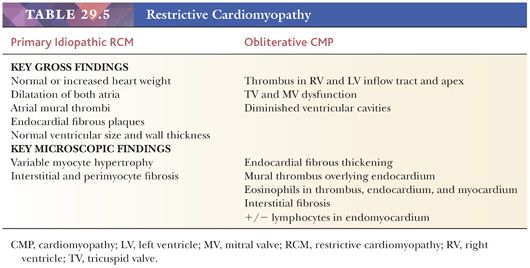
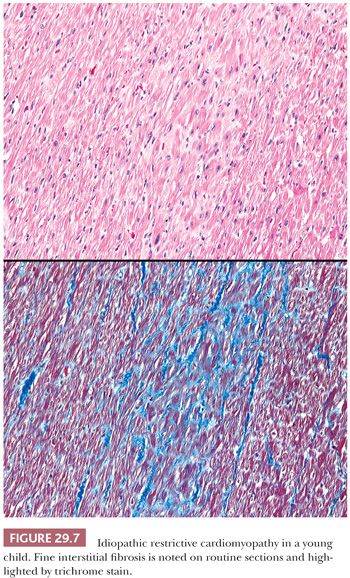
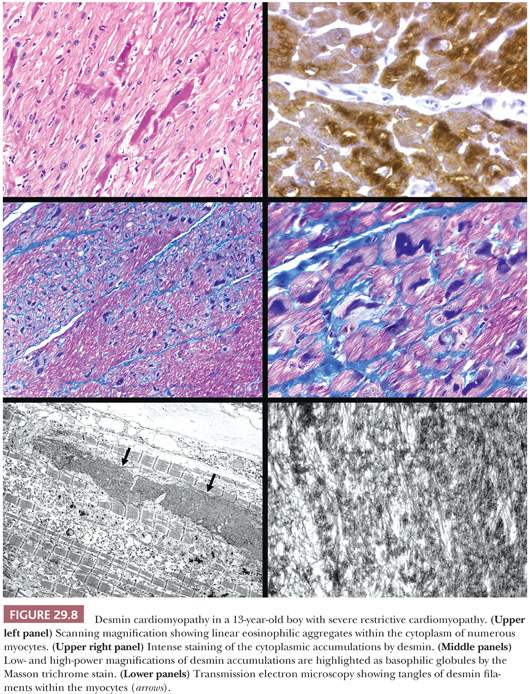
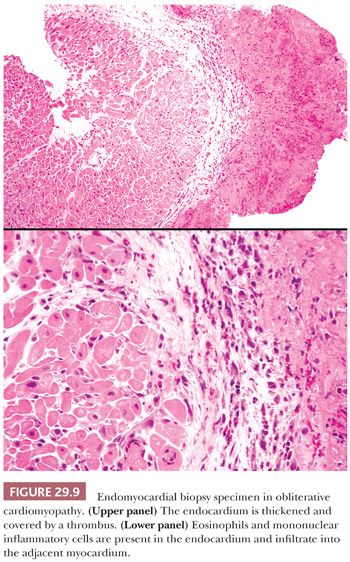
Two forms of the primary disorder have been described (Table 29.5) (114). Primary idiopathic restrictive cardiomyopathy is generally a sporadic disorder that affects children and adults and can be associated with a skeletal myopathy (115–118). Familial forms have also been reported including troponinopathies and desminopathies (74). In many children, there is progression from diastolic dysfunction to systolic heart failure requiring transplantation (48). At transplantation, the heart weight is normal or increased, and the ventricular chamber dimensions and wall thickness and epicardium are normal. Endocardial fibrous plaques are sometimes observed. Biatrial dilatation is present, and thrombi can be found in the appendages. Microscopic sections display variable degrees of myocyte hypertrophy and interstitial fibrosis both in distribution and severity (Fig. 29.7). Desmin cardiomyopathy is characterized by the presence of intracellular filamentous inclusions that exhibit immunoreactivity against desmin (Fig. 29.8). The second type of restrictive cardiomyopathy is also called obliterative cardiomyopathy. It is characterized by endomyocardial abnormalities and consists of an acute form (Loffler endocarditis) and a chronic form (endomyocardial fibrosis [EMF]). The endocardium of the inflow tracts and apices are covered by thrombus. The EMB shows thrombus overlying granulation tissue attached to the endocardium as irregular aggregates (Fig. 29.9). Eosinophils are admixed with the thrombus and may extend into the underlying myocardium. In the later stages, the number of eosinophils diminishes, and the endocardial plaques are thick and fibrotic. This resembles the findings in EMF. Inflow tracts and papillary muscles are covered by collagenous fibrosis and calcifications with reduction in the chamber size. Lymphocytic infiltrates and interstitial fibrosis may be found in the myocardium.
Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia
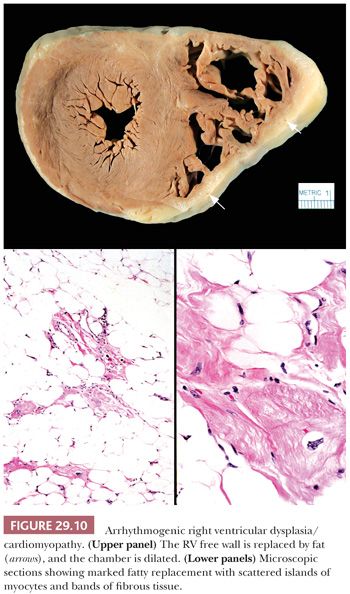
Arrhythmogenic RV cardiomyopathy/dysplasia (ARVC/D) is a recent addition to the primary cardiomyopathy group (72,73). It is an uncommon disorder affecting adolescents and young adults, with an estimated prevalence of 1 in 2500 to 1 in 5000 in the general population, and is more common in parts of Europe (estimated prevalence of 20:5000) than North America (119). The clinical presentation typically is exercise-induced ventricular arrhythmias and sudden death. The clinical diagnosis can be difficult, and a series of major and minor criteria have been established (120). Asimaki and colleagues (121) recently demonstrated reduced immunohistochemical staining for plakoglobin at the intercalated disks in ARVC. An autosomal dominant pattern of inheritance has been reported in the majority of cases, and mutations in genes encoding desmosomal components like plakophilin-2, desmoglein-2, plakoglobin, and desmoplakin have been identified (48,120,122–124). In its classic form, ARVC/D affects the RV either segmentally or globally with replacement of the myocardium by adipose tissue and fibrous tissue. The RV inflow tract, apex, and outflow tract are involved, and at the time of transplantation or autopsy, the RV is dilated and the free wall is attenuated (Fig. 29.10). In some cases, collections of mononuclear inflammatory cells are found within the adipose tissue. The LV can also be affected but usually in conjunction with RV disease. More recently, left dominant and biventricular forms of the disorder have been reported (123). The role of the EMB in the diagnosis of ARVC/D is limited, and the findings must always be interpreted and reported with the clinical and imaging findings. The RV anterior free wall and apex are sites of fatty infiltration in a variety of conditions including normal hearts and in DCM (125).
Left Ventricular Noncompaction
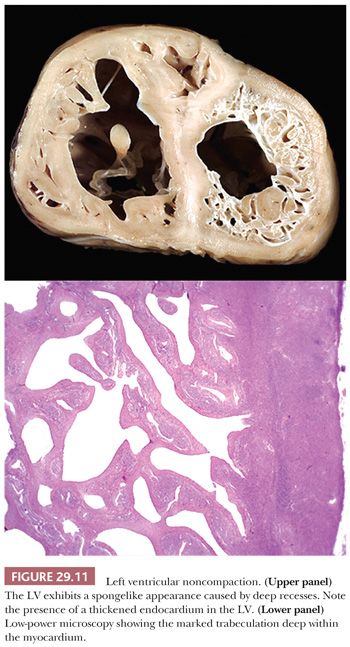
LV noncompaction (LVNC) or “spongy myocardium” is another primary cardiomyopathy that was first included in the 1995 WHO classification. In most cases, it is a congenital disorder that features prominent trabeculations of the LV and deep intertrabecular recesses or sinusoids resulting in its “spongy” appearance (Fig. 29.11). The sinusoids are in direct communication with the ventricular cavity (126,127). The lesion is segmental and shows a predilection for the apical region and mid-LV cavity. Burke and colleagues have reported other patterns including prominent ventricular trabeculations with deep recesses and a dysplastic form with myocardial thinning and exuberant trabeculations (128). Microscopic sectioning shows fibroelastotic thickening of the endocardium, anastomosing endocardial trabeculations, and interstitial fibrosis (128). Foci of ischemic necrosis and vacuolization can be found in the subendocardial zone and in the thickened trabeculae. LVNC occurs from arrested embryologic development of the ventricle (72). It may be a solitary abnormality or seen in association with other structural abnormalities such as pulmonary atresia, atrial/ventricular septal defects, patent ductus arteriosus, or as part of Barth or Noonan syndromes. Both sarcomeric and nonsarcomeric gene mutations have been reported (48,74,129,130). Recently, Luxan and colleagues (131) reported mutations in the Notch pathway regulator MIB1 that prevent ventricular chamber development. Some patients require heart transplantation for intractable heart failure or arrhythmias, and patients are at increased risk for systemic thromboembolic events. The clinical criteria for establishing the diagnosis are evolving but currently are based on echocardiographic and cardiac MRI findings. The diagnosis requires cytoarchitectural assessment of both macroscopic and microscopic examination and therefore cannot be made by EMB.
OTHER TYPES OF PRIMARY CARDIOMYOPATHY
In the recent AHA classification of primary cardiomyopathies, several disorders have been included that the pathologist should be aware of but that do not have specific or recognized histopathologic patterns (72). On occasion, an EMB will be obtained to evaluate an unexplained arrhythmia or conduction defect. In these cases, the pathologist should pay careful attention to the distribution and severity of fibrosis and look for inflammation or granulomas. Molecular studies have identified mutations in several genes responsible for ion channel proteins, and these have been grouped into ion channelopathies. The number of disorders continues to expand and includes long QT syndrome, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia, short QT syndrome, and idiopathic ventricular fibrillation (132). Frustaci et al. (133) showed a spectrum of histopathologic changes in right and left EMBs in patients with the clinical presentation of Brugada syndrome and normal cardiac structure and function on noninvasive workup. These changes included lymphocytic myocarditis, fibrofatty change, and hypertrophy and fibrosis of DCM. Many investigators now promote the concept of the “molecular autopsy” using genetic testing to further investigate these disorders in patients who experience sudden death (59,134). We expect that many of the cardiomyopathies that are currently unclassified will be clarified by molecular genetic studies in the future.
SECONDARY CARDIOMYOPATHIES
The term secondary cardiomyopathy has been proposed to replace specific cardiomyopathy and specific heart muscle disorder of previous classifications. As such, it is defined as a disorder that “shows pathological myocardial involvement as part of a large number and variety of generalized systemic (multiorgan) disorders” (72). This group includes infiltrative and storage disorders; drug, radiation, and chemical toxicities; endocrinopathies; neuromuscular disorders; nutritional and electrolyte disturbances; and autoimmune diseases (Table 29.2). The role of the EMB is twofold: (a) to establish the diagnosis in those disorders with characteristic morphologic findings (e.g., amyloidosis, glycogen storage disorder, Fabry disease, anthracycline toxicity, and hemochromatosis) and (b) to identify treatable or reversible causes of myocardial dysfunction (e.g., catecholamine effect, myocarditis, and neoplasia). The more common diseases are discussed herein.
Amyloidosis
In our experience, the majority of patients with cardiac amyloidosis present in one or more of the following manners: (a) restrictive physiology in the absence of constrictive pericarditis, (b) congestive heart failure that remains refractory to maximal medical therapy, (c) low-voltage changes on electrocardiogram in association with marked LV hypertrophy, or (d) myocardial dysfunction in the setting of known plasma cell dyscrasia or connective tissue disorder. With the exception of isolated senile atrial amyloidosis, which is characterized by beaded subendocardial deposits in the left atrial myocardium found incidentally at autopsy, four types of amyloidosis are associated with cardiac or multiorgan symptoms (135–138). The most common type is immunoglobulin-associated amyloidosis (AL type), which occurs in primary amyloidosis, multiple myeloma, and other types of plasma cell dyscrasia and is characterized by deposition of immunoglobulin light chain. This form is associated with a poor prognosis, and therapy is directed at heart failure management and chemotherapy for the underlying malignancy. Heart transplant followed by myeloablative chemotherapy and autologous stem cell transplantation has been used successfully for carefully selected patients (139–141). Familial or hereditary amyloidosis (ATTR type) is an autosomal dominant disease caused by 1 of 80 different mutations of the transthyretin protein. The mutant TTR protein deposits in the heart and in nerves can cause heart failure and peripheral and autonomic neuropathy. The long-term prognosis is better than AL type, and transplantation is the definitive treatment. Systemic senile amyloidosis is characterized by deposition of wild-type transthyretin and occurs in middle-aged and older men. Its clinical progression and outcome are much better than AL type; unlike AL type, cardiac transplantation is not a contraindication for advanced cardiac involvement. Cardiac involvement in secondary amyloidosis (AA type) is uncommon, and symptoms are minor. Deposition of the nonimmunoglobulin protein A occurs in the setting of chronic infections and autoimmune disorders such as rheumatoid arthritis.
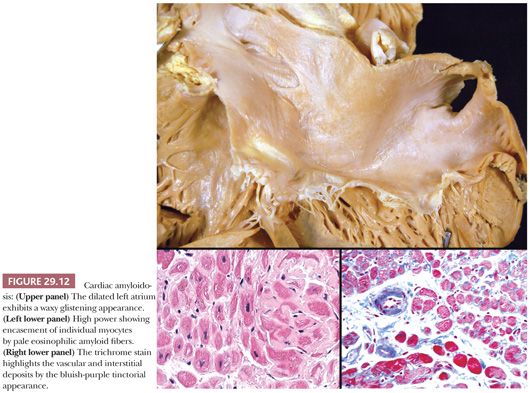
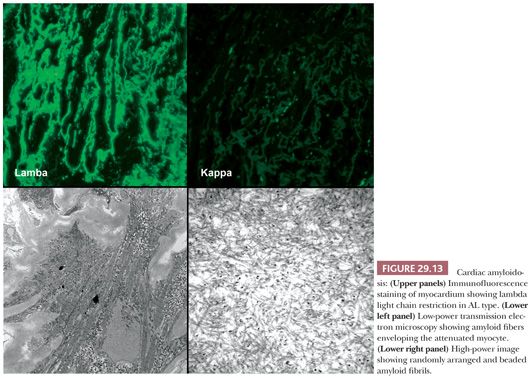
The macroscopic findings in cardiac amyloidosis range from normal ventricles to dilated ventricles with thinned walls (mimicking DCM) to thickened LV walls with small chambers (mimicking HCM). The cut surface is often described as waxy (Fig. 29.12). Involvement of the endocardium, myocardium, pericardium, valves, and epicardial and intramyocardial vessels is common (142). Microscopically, several different patterns are observed. The most common pattern is interstitial and perimyocytic deposition of pale, finely fibrillar eosinophilic material that envelops individual myocytes in a cast-like manner. Attenuated myocytes are frequent and best seen in longitudinally arranged fibers. In our experience, the Masson trichrome stain distinguishes the amyloid fibers from collagen by its gray-blue tinctorial appearance (Fig. 29.12). In addition, collagen shows artifactual retraction from the adjacent myocyte, whereas amyloid encases the myocytes. Coalescence of interstitial deposits to form nodular aggregates is a common feature. The walls of small intramyocardial arteries and the subendocardial tissues are replaced or expanded by amyloid deposits. Classically, in 10-μm sections stained with Congo red, amyloid shows orangiophilic staining under light microscopy and apple green birefringence under polarized light microscopy. Other histochemical stains, such as sulfated Alcian blue, stain amyloid deposits green (143). Ultrastructurally, the perimyocytic and intramural vascular deposits are found, and at high magnification, the fibrils are 7 to 10 nm, randomly arranged, beaded, and nonbranching (Fig. 29.13). In most centers, frozen section immunohistochemistry or immunofluorescence is required to subclassify the different types of amyloid. Because this has important therapeutic and prognostic implications, clinicians should routinely submit tissue in Zeus medium or normal saline as part of the biopsy procedure when amyloid is a diagnostic consideration. Mass spectrometry can also be used to subtype amyloid deposits from formalin-fixed paraffin-embedded samples (144,145).
Glycogen Storage Disease
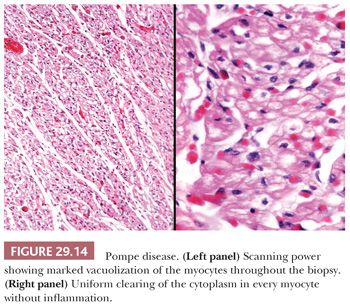
Glycogen storage disorder is a group of inherited enzymatic deficiencies responsible for the synthesis or utilization of glycogen. Of the 12 types currently reported, types II (Pompe), III (Forbe), and IV (Andersen) have cardiac manifestations (146,147). Pompe disease is an autosomal recessive disorder caused by a deficiency of acid maltase (acid alpha-glucosidase). In the infantile form, hepatomegaly, failure to thrive, hypotonia, macroglossia, and massive cardiomegaly occur as a result of accumulations of glycogen within hepatocytes, skeletal muscle, and myocytes. The heart is enlarged and simulates HCM, although DCM and restrictive patterns have also been reported. Endocardial fibroelastosis is observed in up to 20% of cases (146). Histologic sections show enlarged myocytes with vacuolated cytoplasm filled with periodic acid-Schiff (PAS)–positive glycogen (Fig. 29.14). The key feature distinguishing Pompe disease from the vacuolization of normal myocytes is the uniformly massive and diffuse vacuolization of all the myocytes in Pompe disease. Currently, enzyme replacement therapy using recombinant human enzyme is being evaluated in clinical trials (148).
Fabry Disease
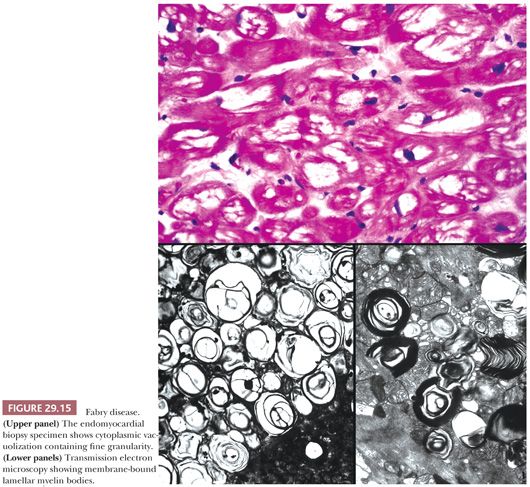
Fabry disease is another example of a storage disorder and is caused by a deficiency of the alpha-galactosidase A enzyme. This X-linked disorder results in glycophospholipid accumulations in the skin, kidneys, liver, pancreas, nervous system, and heart. Cardiac involvement affects endothelial cells, myocytes, valvular structures, and the conduction system, causing ventricular hypertrophy and heart failure, arrhythmias, and conduction defects. Marked LV hypertrophy with either concentric or asymmetric patterns mimics HCM both macroscopically and functionally. In EMB sections, the myocytes and endothelial cells are diffusely vacuolated with pinpoint weakly positive predigested PAS granules within these spaces (Fig. 29.15). The diagnosis is confirmed at the ultrastructural level by the presence of membrane-bound electron-dense lamellar myelin (“zebra”) bodies. Valvular deposition can produce stenotic or regurgitant valves, and severe coronary artery disease has been reported (149,150). Recently, enzyme replacement therapy using recombinant human alpha-galactosidase A enzyme has shown tremendous promise for stabilizing and improving symptoms (151).
Chloroquine cardiotoxicity can mimic the clinical and light microscopy features of Fabry disease (152–154). This potent antimalarial drug is also used in the treatment of sarcoidosis, collagen vascular disorders such as rheumatoid arthritis, and cutaneous discoid lupus. Diffuse myocyte vacuolization is observed. At the ultrastructural level, electron-dense myelin figures and curvilinear bodies are found within myocytes; the latter finding is not seen in Fabry disease.
Hemochromatosis
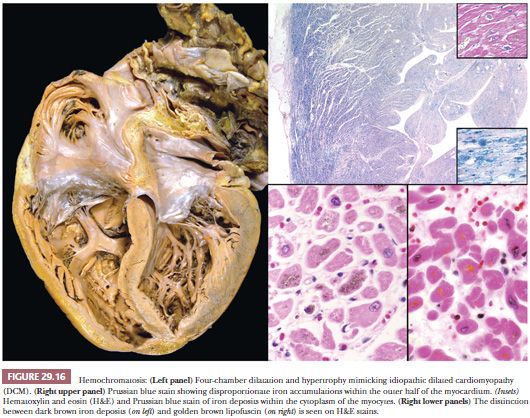
Hemochromatosis is one of the most common inheritable metabolic disorders, with a reported prevalence of 1:200 to 1:400. It is characterized by increased iron absorption and deposition in the liver, heart, pituitary gland, pancreas, joints, adrenal glands, and skin. The pattern of inheritance is autosomal recessive, but the penetrance is variable, accounting for the difficulty in diagnosis (155). The most common mutations in the HFE gene on the short arm of chromosome 6 are C282y, H63D, and S65C. A smaller number of cases are linked to mutations of genes coding SLC11A3 and transferrin receptor 2 (156,157). The cardiac manifestations reflect the effects of iron deposition in myocytes and include congestive heart failure, conduction defects, and arrhythmias. Systolic pump failure causing a DCM is the most common pattern, although cases presenting with a restrictive physiology are reported (Fig. 29.16). Microscopically, iron is deposited in myocytes throughout the myocardium, although a preferential accentuation in the outer half of the myocardium is often observed. The brown deposits are typically perinuclear and are highlighted by the Prussian blue stain. Lipofuscin pigment is distinguished from iron particles by the small uniform golden yellow appearance and lack of staining on iron stains. In advanced cases, interstitial fibrosis and other features of remodeling are present. After transplantation, repetitive phlebotomy is required to prevent recurrence of the disease (158).
Pheochromocytoma and Other Endocrinopathies
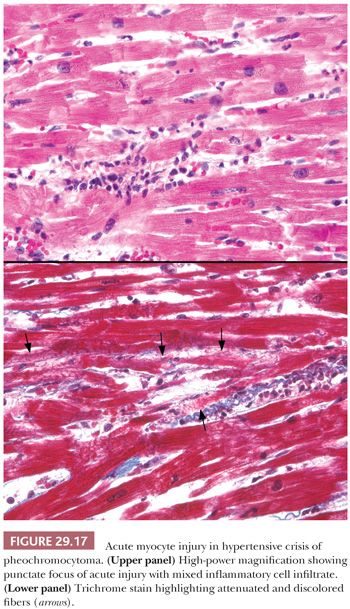
Endocrine disorders may be associated with a variety of cardiomyopathic conditions. Thyroid disorders can produce DCM that is reversible with appropriate interventions (159). Acromegaly produces pronounced LV hypertrophy, interstitial fibrosis, and lymphocytic myocarditis and is influenced by multiple factors such as growth hormone effect, hypertension, long-standing arrhythmias, and vascular changes (160,161). Pheochromocytomas may produce acute or chronic changes in the heart. The release of large amounts of catecholamine produces constriction of small “end vessels” in the myocardium, leading to “microinfarcts” in the myocardium. These lesions are composed of necrotic fibers admixed with scant acute and chronic inflammatory cells (Fig. 29.17) (162). Repetitive and episodic hormone release may lead to arrhythmia- or stress-induced injury that progresses to a DCM. The histopathologic findings are nonspecific, and the diagnosis requires clinical screening (163).
Anthracycline Cardiotoxicity
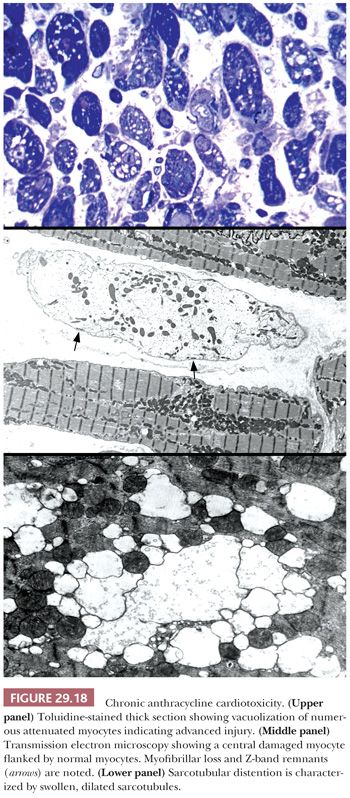
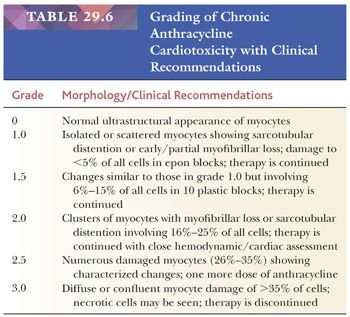
The most common anthracycline antibiotics used in the treatment of solid tumors and hematolymphoid malignancies are doxorubicin and daunorubicin. Their efficacy is limited by their cardiotoxicity—a complication that has been recognized for more than 30 years. Both acute, early-onset chronic progression and late-onset chronic forms have been described (164,165). The late-onset chronic form can present many years or decades after treatment and is dose dependent, cumulative, and progressive. Congestive heart failure or arrhythmias are common and may be precipitated by other clinical events such as infection, pregnancy, or surgery (166). As previously discussed, the morphologic assessment of chronic anthracycline toxicity requires special tissue handling. All the issue is fixed and processed in a glutaraldehyde solution for transmission electron microscopy. Using the Billingham criteria, a minimum of 10 epon plastic blocks is required to confidently assess a numeric grade (167). One-micron sections are cut and stained with toluidine blue stain and assessed under light microscopy for distribution and extent of cellular injury. Myocytes with myofibrillar loss appear shrunken with homogeneous pale cytoplasm, and sarcotubular dilatation is depicted by cytoplasmic vacuolization (Fig. 29.18). At the ultrastructural level, myofibrillar loss with partial or complete loss of fibrils and retention of Z-band remnants along the cytoplasmic membrane is more common than the swelling and coalescence of vacuolar change to form sarcotubular distention. The grading of cardiotoxicity ranges from 0 to 3, and each grade is associated with specific therapeutic recommendations (Table 29.6) (164). Approaches to reducing the incidence of cardiotoxicity include the use of liposomal formulations of the anthracyclines and cardioprotective drugs such as dexrazoxane (168,169).
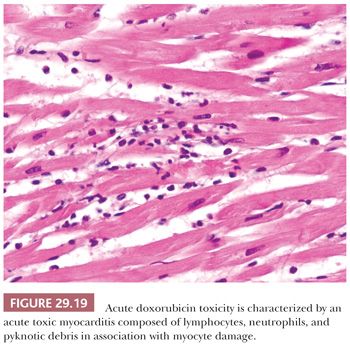
An uncommon form of anthracycline toxicity develops after the first dose or within the first week of therapy. Patients present with signs and symptoms of acute pericarditis/myocarditis. The myocardial lesions are typically patchy and are composed of a mixture of neutrophils and lymphocytes in association with myocyte damage (Fig. 29.19) (170).
Cardiotoxicity is reported with other chemotherapeutic drugs (171,172). In many cases, the changes are functional rather than morphologic. Alkylating agents such as busulfan and cyclophosphamide can cause EMF and pericarditis/myocarditis, respectively. DCM is reported in the setting of human immunodeficiency virus (HIV) infection and may be related to the viropathic effects of HIV or the antiviral drugs such as zidovudine (azidothymidine [AZT]) (173).
INFLAMMATORY CARDIOMYOPATHY (MYOCARDITIS)
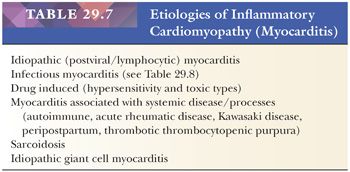
In patients with recent onset (<3 months in duration) of unexplained congestive heart failure with normal-sized or dilated heart and/or ventricular arrhythmias, the EMB is used to evaluate for myocarditis (78). This category includes numerous etiologies of myocarditis (Table 29.7) (174,175). For the purpose of this chapter, we will present the types of myocarditis that may be encountered in surgical pathology and refer the reader to other monographs for more comprehensive discussions (176–178).
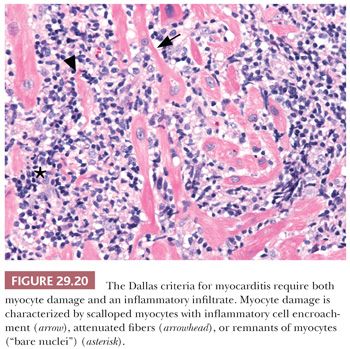
In 1984, a consensus definition and classification of myocarditis was proposed (179). Myocarditis was defined as a myocardial process characterized by the presence of both an inflammatory infiltrate and myocyte damage or necrosis not typical of the myocardial damage of ischemic heart disease. In active myocarditis, both inflammation and myocyte damage are present. The composition of the cellular infiltrate delineates the specific type of myocarditis (e.g., lymphocytic, eosinophilic, neutrophilic, giant cell, granulomatous, or mixed cell types). The distribution of inflammatory infiltrate may be focal, confluent, or diffuse; the severity ranges from mild to severe. The most difficult challenge is the determination of myocyte damage in the biopsy specimen. In our experience, florid myocytolysis and necrosis are not common biopsy patterns. Myocyte damage is characterized by the presence of mononuclear cells that cause encroachment or scalloping of the sarcolemmal membrane of myocytes, fragmentation of myocytes with remnants of cytoplasm or “bare nuclei,” architectural displacement or distortion of myocytes by inflammatory cells, or partial replacement of myocytes by inflammatory cells (Fig. 29.20). The liberal use of leveled sections and Masson trichrome are helpful in difficult cases because damaged myocytes display a basophilic tinctorial quality. Finally, the presence or absence of fibrosis should be noted for reference to changes in subsequent biopsies and the potential development of DCM. Interstitial, perivascular, and endocardial patterns can be seen. Borderline myocarditis is defined by the presence of limited inflammatory cell infiltrates and the absence of definitive myocyte damage. The phrase no evidence of myocarditis is used when neither diagnostic feature is present. Deeper sectioning of the paraffin blocks should also be considered before this diagnosis is rendered because the inflammatory process may be patchy in distribution. If myocarditis is absent, attention should be focused on the presence of other myocardial disorders such as myocyte hypertrophy and interstitial fibrosis in the setting of DCM. Connective tissue stains and stains for amyloidosis and hemochromatosis should then be routinely obtained. Follow-up biopsies after therapeutic intervention are less commonly performed today. Possible diagnostic observations include ongoing or persistent myocarditis, resolving or healing myocarditis, and complete resolution of inflammation and damage. We have observed cases of recurrent myocarditis after rapid tapering of immunosuppressive therapy. Furthermore, the development of myocyte hypertrophy and interstitial fibrosis should be noted in follow-up biopsies and correlated with physiologic and imaging findings because progression to DCM has been observed (180).
Lymphocytic Myocarditis
In developed countries, viral agents are thought to be the primary cause of lymphocytic myocarditis. Historic, clinical, and experimental evidence has identified enteroviruses such as coxsackie B virus, echovirus, and adenovirus as likely causes (177,181). More recently, the role of parvovirus B19, human herpesvirus 6, and other viruses in myocarditis and DCM has been postulated, but there remains a degree of controversy (182–185). Possible mechanisms include postviral autoimmunity mechanisms; direct viral cytopathic injury; induction of viral-specific immune response through mediators such as interleukin (IL)-1, IL-2, IL-6, tumor necrosis factor (TNF), interferon, and nitrous oxide; and viral-mediated endothelial injury with intimal proliferation and ischemic sequelae. In many clinical cases, however, a direct causative link is not established, and these cases are classified as idiopathic myocarditis. Other terms that have been used include acute myocarditis to reflect the clinical onset of symptoms and absence of fibrosis in the biopsy specimen or rapidly progressive or fulminant myocarditis in cases of multifocal damage and extensive injury.
The incidence and natural history of idiopathic myocarditis remain largely unknown. Discrepancies between clinically suspected cases and EMB findings are well recognized, and the prevalence of myocarditis using the EMB as the gold standard is less than 10% (186).
The macroscopic findings in florid myocarditis typically range from normal cardiac configurations to four-chamber dilatation and cardiac enlargement. The papillary muscles and trabeculae carneae are often flattened, and the myocardium appears pale and flabby. Thrombi within atrial appendages or along ventricular endocardial surfaces are uncommon. The cut surface of the myocardium is usually pale, and foci of hemorrhage or hemorrhagic necrosis are found. Many cases have fibrinous pericarditis and exudative effusions.
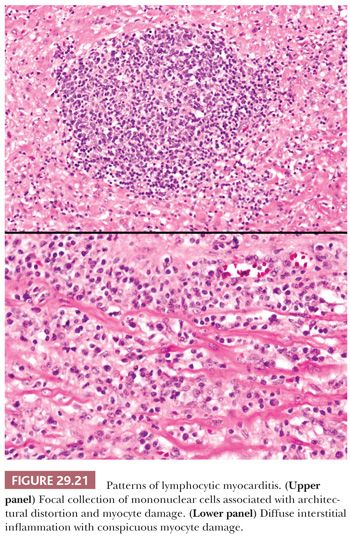
The resemblance of this type of myocarditis and acute cellular rejection of the cardiac allograft has been previously noted (187). In many cases, the infiltrates are sparse and are predominantly lymphocytic in nature. In ful minant cases, myocyte damage or necrosis is conspicuous (Fig. 29.21). Architectural patterns include focal, multifocal, or diffuse interstitial infiltrates. Interstitial widening by tissue edema and inflammation is seen. The patterns of confluent myocyte damage and necrosis are similar in biopsies of adult and pediatric patients. The majority of lymphocytes are CD3+ T cells with both helper and suppressor subtypes. Macrophages and natural killer cells are also present, but B cells are infrequent or absent.
There are several common histopathologic lesions that can be mistaken for lymphocytic myocarditis, and many are included in Table 29.7. The average number of lymphocytes in normal myocardial tissue is thought to be less than 5.0 per high-power field (188). Tazelaar and Billingham (189) examined EMBs obtained from 86 young disease-free cardiac transplantation donors at the time of transplantation. Foci composed of at least five mononuclear inflammatory cells were found in 9.3% of cases. Other types of interstitial cells normally found within the myocardium that can be confused with lymphocytes are endothelial cells, smooth muscle cells, pericytes, fibroblasts, and mast cells. Hill and Swanson (190) reported the presence of extramedullary hematopoietic cells including immature erythroid and myeloid precursors in healing infarcts of ischemic and cardiomyopathic hearts and areas of fibrosis in congenital defects. Foci of mononuclear leukocytic cells are found in the interstitial tissues and within fibrosis in most cases of DCM but are not associated with myocyte damage (91). Vasopressor/catecholamine-damaged myocytes appear fragmented and hypereosinophilic and are surrounded by a sparse mixed inflammatory cell infiltrate, which is similar to the lesions seen with pheochromocytomas (162). The distribution of the lesions near or around small intramyocardial arteries and the mixed nature of the infiltrate are important diagnostic clues. The trichrome stain highlights the necrotic myocytes. The damaged myocytes may undergo punctate calcification and mimic infectious myocarditis such as toxoplasmosis. Neoplastic infiltrates are uncommon findings on EMBs. Their atypical cytologic features and the absence of necrosis and fibrosis characterize hematolymphoid malignancies such as leukemia and lymphoma. Immunophenotypic and molecular studies are helpful to confirm the clonality of these processes and to distinguish them from myocarditis.
Infectious Myocarditis
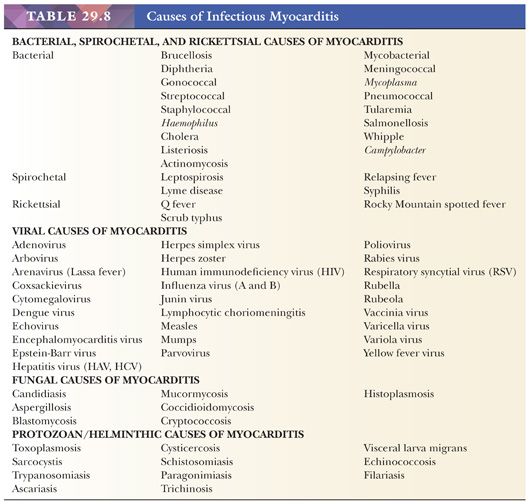
In developed countries, infectious causes of heart muscle inflammation are uncommon in immunocompetent individuals. Patients with acquired immunodeficiency syndrome (AIDS), transplantation-associated immunosuppression to prevent allograft rejection, and advanced stages of malignancy are susceptible to a variety of bacterial, viral, fungal, protozoan, and rickettsial infections. In many developing countries, these remain a significant cause of morbidity and mortality, and cardiac involvement is frequently observed. Careful clinical history; cultures; serologies; and histochemical, immunohistochemical, and molecular studies are usually necessary to establish the diagnosis. More detailed descriptions of these different infections are found in other monographs (Table 29.8) (176).
Drug-Related Myocarditis
Drug-induced myocardial dysfunction remains a significant clinical problem, and the list of implicated drugs continues to expand. Five patterns are recognized: (a) hypersensitivity myocarditis, (b) toxic myocarditis, (c) endocardial fibrosis (e.g., ergotamine tartrate, methysergide, pergolide mesylate, phentermine/fenfluramine), (d) drug-induced cardiomyopathy (e.g., anthracycline, chloroquine), and (e) giant cell myocarditis (191–194).
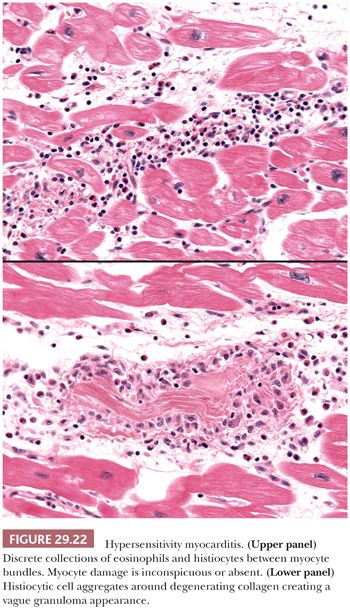
Hypersensitivity Myocarditis. Hypersensitivity myocarditis is the most common form of acute drug-related myocardial injury. More than 40 drugs have been identified, but the majority of cases are caused by antibiotics, diuretics, and antihypertensive drugs (195). It is also observed in 7% of patients undergoing cardiac transplantation and is likely related to prolonged dobutamine infusion (196–200). Clinical signs can include rash, fever, peripheral eosinophilia, and occasionally arrhythmias, sudden death, and congestive heart failure. It is not dose dependent and may occur at any time during drug administration. The histopathologic features include temporally uniform lesions distributed in the subendocardial, perivascular, and interstitial tissues between bundles of myocytes. The predominant inflammatory cells are eosinophils, but variable numbers of histiocytes and scattered lymphocytes are also found (Fig. 29.22). Myocyte necrosis is usually absent or very focal except in severe cases. Necrotizing vasculitis is not found, but infiltration of vessel walls by eosinophils is common (201). Collections of histiocytes centered on degenerated collagen bundles form ill-defined granulomas in up to 25% of cases, but fibrinoid necrosis, well-formed aggregates of epithelioid histiocytes (“hard granulomas”), multinucleated giant cells, interstitial fibrosis, and hemorrhage are absent in our experience. The lymphocytes show a T-cell phenotype and sparse or absent B cells. The absence of diffuse myocardial necrosis and giant cells distinguishes hypersensitivity myocarditis from drug-induced giant cell myocarditis (192). Acute necrotizing eosinophilic myocarditis differs from classic hypersensitivity myocarditis by the presence of extensive necrosis, dense interstitial inflammatory cell infiltrates, and absence of systemic allergic symptoms (202,203).
Toxic Myocarditis. Toxic myocarditis is an uncommon form of myocarditis and is characterized by direct myocyte cytotoxicity. Causative agents include antineoplastic agents such as cyclophosphamide and anthracyclines, catecholamines, cocaine, arsenicals, fluorouracil, lithium compounds, and antihypertensives (191,195). In distinction to hypersensitivity myocarditis, it is usually dose dependent, and the lesions may persist or progress after the cessation of the drug. The pathologic features reflect the cellular response to myocyte damage. The lesions are focal and temporally heterogeneous, reflecting the episodic or cumulative mechanism of injury. Some lesions in the biopsy sample may be acute, whereas others may be in the reparative phases. Fibrosis is not uncommon. The inflammatory infiltrates are polymorphous with lymphocytes, plasma cells, and neutrophils, but eosinophils are rare or absent. A classic example is the acute form of doxorubicin cardiotoxicity presenting as acute myocarditis (Fig. 29.19).
Myocarditis Associated with Systemic Processes
Myocarditis has been reported in several systemic illnesses and in the peripartum period. Many of these are examples of immune-mediated myocarditis and exhibit lymphocytic myocarditis.
Collagen Vascular Diseases. Myocarditis is reported in connective tissue diseases such as systemic lupus erythematosus (SLE), systemic sclerosis, polyarteritis nodosa, rheumatoid arthritis, polymyositis/dermatomyositis, thrombotic thrombocytopenic purpura, Wegener granulomatosis, and rarely, ankylosing spondylitis and mixed connective tissue disease (204,205). SLE, rheumatoid arthritis, and polymyositis/dermatomyositis are most commonly associated with myocarditis. The morphologic features on EMB or in postmortem material are lymphocytic myocarditis similar to the idiopathic (postviral) type of myocarditis. This emphasizes the importance of comprehensive clinical information in the evaluation of these cases. In SLE, fibrinoid vasculitis may also be observed in the small intramyocardial arteries in the biopsy. Immunofluorescence studies may demonstrate immunoglobulin, complement, and fibrinogen deposition, indicating an immune complex–mediated form of myocarditis. Immunosuppressive therapy remains the mainstay of treatment. Drug-related toxic myocarditis should be considered in the differential diagnosis, particularly in SLE patients receiving quinidine-based therapy.
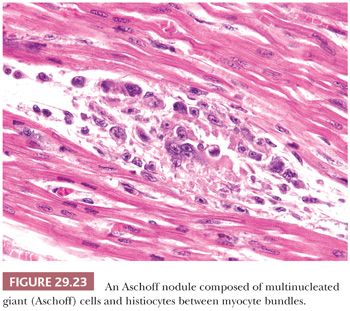
Acute Rheumatic Fever. Rheumatic fever remains a significant cause of cardiac morbidity and mortality in underdeveloped countries (206). It is a sequel to group A streptococcal pharyngitis and arises as an autoimmune response to extracellular or somatic bacterial antigens that share similar epitopes in human tissues. Cardiac involvement occurs in up to 55% of patients and is characterized by a pancarditis. The diagnosis of rheumatic myocarditis has been made on EMB, at the time of transplantation and at autopsy (207,208). The myocardial lesions consist of nonspecific lymphocytic myocarditis and Aschoff nodules. The latter may be found in the valves, endocardium, myocardium, pericardium, and conduction system and are pathognomonic of acute rheumatic fever. They represent oval collections of histiocytes, lymphocytes, plasma cells, and giant cells (Aschoff cells) in the interstitium adjacent to small blood vessels (Fig. 29.23). This “granulomatous stage” of Aschoff nodules arises 1 to 2 months after the onset of clinical symptoms and develops within or near foci of fibrinoid necrosis. They are eventually replaced by collagenous scar tissue.
Peripartum Myocarditis/Cardiomyopathy. Peripartum myocarditis/cardiomyopathy is defined as myocardial dysfunction occurring during the third trimester of pregnancy or in the first 5 or 6 months postpartum (209,210). Possible etiologies include viral infection, nutritional deficiencies, small vessel coronary disease, and immunologic interactions to fetal and myometrial antigens. Lymphocytic myocarditis is reported in 5% to 30% of cases. DCM has been occasionally found in association with myocarditis. Whether postpartum myocarditis/cardiomyopathy is a distinct entity from idiopathic DCM or idiopathic lymphocytic myocarditis remains unknown.
Sarcoidosis
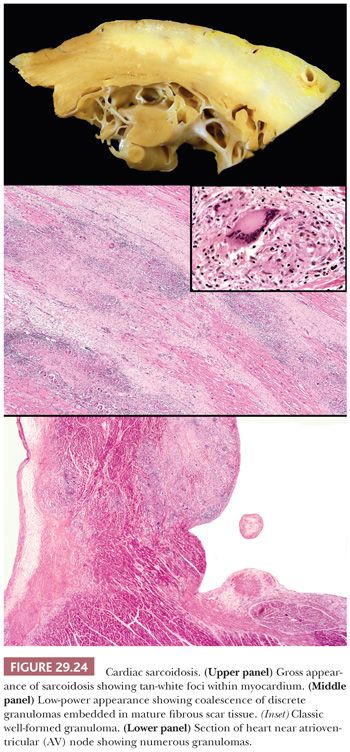
Cardiac involvement in sarcoidosis occurs in 25% to 60% of patients but remains subclinical in the vast majority of cases. Isolated cardiac involvement in the absence of systemic disease is found in a minority of cases. Cardiac dysfunction manifests as arrhythmias, particularly ventricular types; conduction disturbances with high degrees of atrioventricular (AV) block or complete bundle branch block; sudden death; congestive heart failure; papillary muscle dysfunction; acute myocardial infarction (MI)–like syndrome; ventricular aneurysm; or recurring pericardial effusions. In a recent study of cardiac sarcoidosis, left-sided heart failure and syncope were the most common symptoms at the time of hospital presentation. AV block and ventricular tachycardia accounted for more than 75% of arrhythmias, but sudden death occurred in 2% of cases (211). The sensitivity of RV EMB ranges from 20% to 50% because of the patchy distribution of the granulomatous lesions and the preferential localization in the cephalad portion of the interventricular septum, LV free wall, and papillary muscles (212). A negative biopsy does not exclude the diagnosis, and some have advocated institution of immunosuppressive therapy even in the face of a negative biopsy result (213). Corticosteroid therapy is effective in many cases, and cardiac transplantation remains a therapeutic option for patients who fail medical therapy or progress to DCM. Recurrence in the allograft has been reported but is uncommon; treatment with augmented immunosuppression is efficacious (214).
Several histopathologic patterns can be observed on EMBs in cardiac sarcoidosis. These include the classic noncaseating granulomatous inflammation, lymphocytic myocarditis, DCM, and normal myocardium. Diffuse myocardial involvement progresses to myocyte hypertrophy and interstitial fibrosis resembling DCM; in a minority of cases, a restrictive profile is observed. The classic granulomatous pattern is characterized by firm, white nodules forming discrete masses within the interventricular septum, LV free wall, or papillary muscle (Fig. 29.24). These may be confused for metastatic deposits or fibrous tumors. The histopathologic features are similar to extracardiac lesions and consist of noncaseating, well-formed (so-called “hard”) granulomas composed of epithelioid histiocytes and multinucleated giant cells arranged in round or oval aggregates. These can be found as isolated lesions or may coalesce to form larger zones within the myocardium. Endocardial and pericardial involvement is observed in some cases. Scattered around and within the granulomas are mature lymphocytes, but eosinophils are absent or sparse in number. Mature collagenous fibrosis is present and surrounds the granulomas, but active myocyte necrosis is uncommon. The epithelioid histiocytes express CD68, and the infiltrating lymphocytes are almost exclusively T cells with a predominance of CD4+ cells; B cells are rare (215).
The differential diagnosis includes granulomatous and giant cell lesions of the heart. Granulomatous infections are uncommon in immunocompetent patients, but we routinely obtain histochemical stains for fungal and mycobacterial microorganisms. In general, infectious lesions demonstrate necrotizing granulomas. Giant cell myocarditis is characterized by the presence of giant cells, and by definition, granulomas are absent. In hypersensitivity myocarditis, the histiocytic lesions are poorly formed and are centered on collagen fibers. Eosinophils are numerous, but multinucleated giant cells and fibrosis are not found. The granuloma-like lesions of acute rheumatic fever are poorly formed, and the giant cells are generally smaller and do not resemble Langhan type. Foreign body–type giant cells surrounding catheter sheath fragments can be found in biopsies of patients who undergo repeated biopsy procedures. The edge of healing ischemic infarcts can contain giant cells of myogenic origin; lymphocytes and hemosiderin-laden macrophages are seen within the scar tissue. Granulomas are also reported in metabolic disorders such as lipogranulomatosis (Farber disease), oxalosis, and gout; in collagen vascular diseases such as rheumatoid nodules, Wegener granulomatosis, and Churg-Strauss syndrome; and in chronic granulomatous disease of childhood (216).
Idiopathic Giant Cell Myocarditis
Idiopathic giant cell myocarditis (IGCM) is a rare but frequently fatal form of myocarditis that occurs in previously healthy young adults. Clinical onset is abrupt and characterized by rapidly progressive heart failure and/or arrhythmias. Death occurs within weeks or months of onset of symptoms unless aggressive immunosuppression and cardiac transplantation are implemented (217). Twenty percent of patients have an associated autoimmune disorder such as ulcerative colitis, cryofibrinogenemia, rheumatoid arthritis, myasthenia gravis, hyperthyroidism, and hypothyroidism. Other associations include drug hypersensitivity, Wegener granulomatosis, thymoma, sarcoidosis, and infections. This fulminant form of IGCM is distinguished from the indolent isolated atrial variant of GCM that is found incidentally in atrial surgical specimens (218,219).
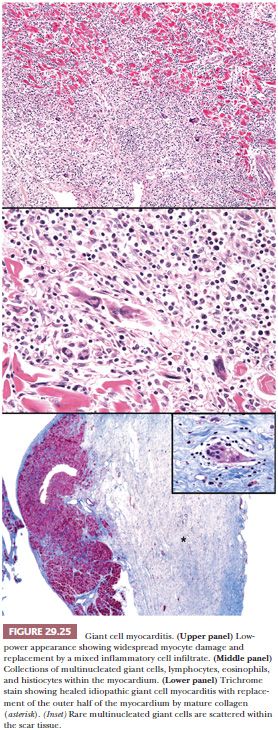
Grossly, confluent or multifocal areas of necrosis are easily observed in the heart. The heart weight is normal or slightly increased. The four chambers of the heart are uniformly involved in most cases. In the late or healed stages of the disease, the ventricular wall may appear thin, but this reflects diffuse scarring and not aneurysmal changes as islands of myocytes are found within the collagenous scar tissue (Fig. 29.25). Endocardial and pericardial involvement has been described, but the process is primarily centered on the myocardium. The microscopic features consist of regions of diffuse, serpiginous necrosis containing multinucleated giant cells, lymphocytes, histiocytes, and eosinophils in the absence of sarcoid-like granulomas. The giant cells are distributed throughout the inflammatory infiltrates often in apposition to the sarcolemmal membranes of necrotic myocytes. They measure up to 90 × 20 μm in size and contain up to 20 nuclei in each cell. The necrotic myocardium is replaced by edematous granulation tissue, and the border between viable and necrotic myocardium is not well delineated. Litovsky et al. (215) have proposed classification of IGCM into acute, healing, and healed phases. The acute or active phase is described earlier and is distinguished by the abundant inflammatory response, loose connective tissue stroma, and numerous giant cells of macrophage origin. In the healing or resolving stage, granulation tissue and immature fibrosis replace the myocardium, and the number of giant cells and inflammatory cells is diminished. In the healed or resolved phase, mature fibrosis is noted with rare or absent giant cells and sparse inflammatory cells (Fig. 29.25). Myocytes are found as islands of single cells or small clusters surrounded by scar tissue. In explanted or postmortem heart specimens, some degree of overlap between the different stages can be seen, suggesting temporal heterogeneity in this disease. This is an important caveat when examining EMB specimens for the purpose of grading the response to immunosuppressive therapy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


