10 Haemopoietic and lymphoreticular system
HAEMOPOIESIS
ANAEMIA
Blood loss
Immediately after acute haemorrhage the haemoglobin level is normal. In the absence of intravenous fluid replacement, there is a slow expansion in plasma volume over the next two to three days. The result is a normochromic, normocytic anaemia. There is also a reticulocytosis, which is maximal at one week, together with a mild neutrophil leucocytosis. Occasionally metamyeloyctes are present in the blood film. Chronic blood loss leads to hypochromic, microcytic, iron deficiency anaemia.
Haemolysis
Classification of anaemia
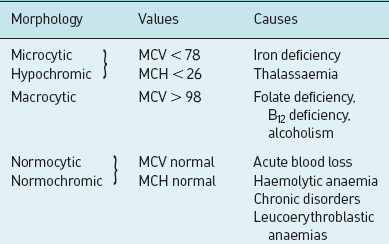
Anaemias may be classified by the morphological appearance of erythrocytes in a stained blood smear. Normocytes are red cells with a normal diameter, microcytes are those with a reduced diameter, and macrocytes are those with an increased diameter. Normochromic is a term applied to normal staining of the red cell with a central area of pallor, while hypochromic indicates reduced staining with a larger central area of pallor. Classification also depends on other criteria. The haematocrit or PCV is expressed as the percentage of packed red cells in relation to the total volume of blood and is normally approximately 45%. Other important parameters in assessing anaemia are:
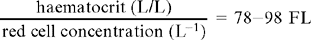

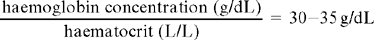
A morphological classification of anaemia is shown in Table 10.1.
Table 10.1 Morphological classifications of anaemia
POLYCYTHAEMIA
Polycythaemia is an increase in the concentration of red cells above the normal level. There is a rise in both total blood volume and PCV; the latter may be as high as 60%. The Hb concentration rises to about 18 g/dL and, because of the increased proportion of erythrocytes, blood viscosity is high. Polycythaemia may be a primary condition, i.e. polycythaemia rubra vera, or may be secondary or relative, or due to inappropriate secretion of erythropoietin (Box 10.1). Polycythaemia, especially the true and secondary forms, increases whole blood viscosity. This leads to sluggish blood flow through the heart, brain and limbs, leading to myocardial infarction, stroke and ischaemic limbs. The spleen is enlarged in about 75% of cases. Haemorrhagic lesions may be a feature especially in the gastrointestinal tract. Peptic ulceration is common in polycythaemia rubra vera, but the reason is unknown.
WHITE BLOOD CELL (LEUCOCYTE)
Changes in white cells in disease
Leucocytosis
Leucocytosis is an increase in the number of circulating white cells. The normal reference range is shown in Table 10.2. It may involve any of the white cells, but a polymorphonuclear leucocytosis is the most common, i.e. neutrophilia (Table 10.3).
Table 10.2 Reference range for white cell concentrations
| Cell | Count (109/L) |
|---|---|
| Total white cell count | 4–11 |
| Neutrophils | 2.0–7.5 |
| Lymphocytes | 1.0–3.0 |
| Monocytes | 0.15–0.6 |
| Eosinophils | 0.05–0.35 |
| Basophils | 0.01–0.10 |
Table 10.3 Causes of leucocytosis
| Cell | Cause |
|---|---|
| Neutrophils | |
| Monocytes | |
| Eosinophils |
PLATELETS
Platelets are discoid non-nucleated granule-containing cells that are formed in the bone marrow by fragmentation of the cytoplasm of megakaryocytes. Their concentration in normal blood is 160–450×109/L. They survive in the circulation for 8–10 days. Platelets are contractile and adhesive cells which are import-ant in haemostasis. They adhere to exposed subendothelial tissues, aggregate, and form a haemostatic plug. Platelets may also take part in the repair process after vascular injury. Platelet-derived growth factor is mitogenic for smooth muscle and fibroblasts; it may also be involved in the development of atheroscler-osis. The function of platelets is discussed further in the section on haemostasis. A reduction in the number of platelets is called thrombocytopaenia (Table 10.5).
Table 10.5 Causes of thrombocytopaenia
| Type | Cause |
|---|---|
| Reduced production | |
| Decreased platelet survival | |
| Immune | |
| Non-immune | |
| Hypersplenism | Sequestration of platelets |
HAEMOSTASIS
Coagulation mechanism
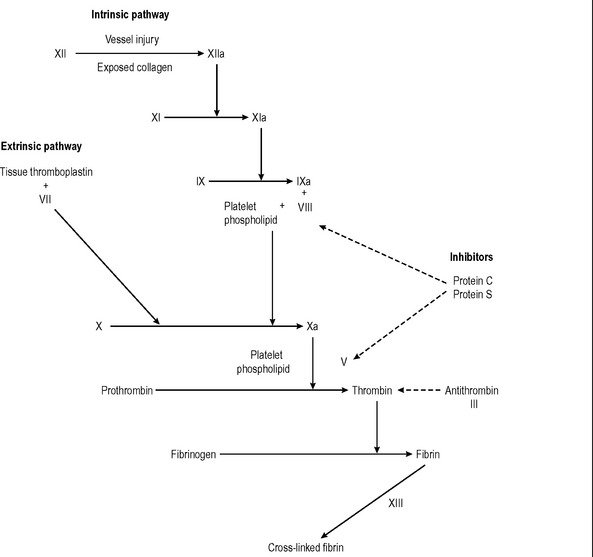
The end-stage of blood coagulation is the conversion of soluble fibrinogen to insoluble fibrin by the protease thrombin. The coagulation mechanism is complex and involves two interacting systems: the intrinsic and extrinsic pathways. Activation of factor X is the result of preceding enzyme reactions in the two pathways. The intrinsic pathway involves normal blood compon-ents; the extrinsic pathway requires tissue thromboplastin released by damaged cells. The pathways are shown in Fig. 10.1. All the soluble coagulation factors are manufactured in the liver with the exception of Factor VIII (endothelium), calcium, platelet factors and thromboplastin.
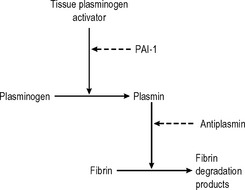
Fibrinolytic system
During the repair process in blood vessels and healing wounds, fibrin is removed by the fibrinolytic system (Fig. 10.2). Fibrin is broken down to soluble fibrin degradation products by plasmin. Plasmin is derived from the inactive precursor plasminogen by the action of plasminogen activators. Tissue plasminogen activator is released from endothelial cells. Control of the activation of plasminogen is provided by plasminogenactivator inhibitor I, which is released by endothelial cells and rapidly inactivates tissue plasminogen activator.