Chapter 8 Haematological disease
Introduction and general aspects




The formation of blood cells (haemopoiesis)
There are two major ancestral cell lines derived from the pluripotential stem cell: lymphocytic and myeloid (non-lymphocytic) cells (Fig. 8.1). The former gives rise to T and B cells. The myeloid stem cell gives rise to the progenitor CFU-GEMM (colony-forming unit, granulocyte–erythrocyte–monocyte–megakaryocyte). The CFU-GEMM can go on to form CFU-GM, CFU-Eo, and CFU-Meg, each of which can produce a particular cell type (i.e. neutrophils, eosinophils and platelets) under appropriate growth conditions. The progenitor cells such as CFU-GEMM cannot be recognized in bone marrow biopsies but are recognized by their ability to form colonies when haemopoietic cells are immobilized in a soft gel matrix. Haemopoiesis is under the control of growth factors and inhibitors, and the microenvironment of the bone marrow also plays a role in its regulation.
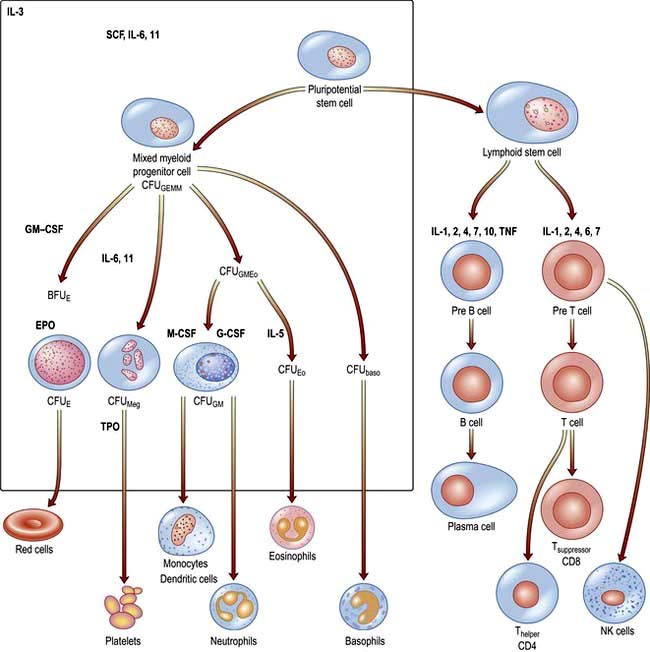
Haemopoietic growth factors
Haemopoietic growth factors are glycoproteins, which regulate the differentiation and proliferation of haemopoietic progenitor cells and the function of mature blood cells. They act on the cytokine-receptor superfamily expressed on haemopoietic cells at various stages of development to maintain the haemopoietic progenitor cells and to stimulate increased production of one or more cell lines in response to stresses such as blood loss and infection (Fig. 8.1).
These haemopoietic growth factors including erythropoietin, interleukin 3 (IL-3), IL-6, -7, -11, -12, β-catenin, stem cell factor (SCF, Steel factor or C-kit ligand) and Fms-tyrosine kinase 3 (Flt3) act via their specific receptor on cell surfaces to stimulate the cytoplasmic janus kinase (JAK) (see p. 25). This major signal transducer activates tyrosine kinase causing gene activation in the cell nucleus. Colony-stimulating factors (CSFs, the prefix indicating the cell type, see Fig. 8.1), as well as interleukins and erythropoietin (EPO) regulate the lineage committed progenitor cells.
Stem cell diseases
The clonal proliferation of bone marrow stem cells leads to diseases including leukaemia (see p. 451), polycythaemia vera (see p. 402), myelofibrosis (see p. 404), paroxysmal nocturnal haemoglobinuria (see p. 401). Failure of stem cell growth leads to aplastic anaemia (see p. 385).
Peripheral blood
Automated cell counters are used to measure the haemoglobin concentration (Hb) and the number and size of red cells, white cells and platelets (Table 8.1). Other indices can be derived from these values. A carefully evaluated blood film is still an essential adjunct to the above, as definitive abnormalities of cells can be seen.
 The mean corpuscular volume (MCV) of red cells is a useful index and is used to classify anaemia (see p. 376).
The mean corpuscular volume (MCV) of red cells is a useful index and is used to classify anaemia (see p. 376).





 C-reactive protein (CRP) is a pentraxin, one of the proteins produced in the acute-phase response. It is synthesized exclusively in the liver and rises within 6 hours of an acute event. The CRP level rises with fever (possibly triggered by IL-1, IL-6 and TNF-α and other cytokines), in inflammatory conditions and after trauma. It follows the clinical state of the patient much more rapidly than the ESR and is unaffected by the level of Hb, but it is less helpful than the ESR or plasma viscosity in monitoring chronic inflammatory diseases. The measurement of CRP is easy and quick to perform using an immunoassay that can be automated. High-sensitivity assays have shown that increased levels may predict future cardiovascular disease (see p. 728).
C-reactive protein (CRP) is a pentraxin, one of the proteins produced in the acute-phase response. It is synthesized exclusively in the liver and rises within 6 hours of an acute event. The CRP level rises with fever (possibly triggered by IL-1, IL-6 and TNF-α and other cytokines), in inflammatory conditions and after trauma. It follows the clinical state of the patient much more rapidly than the ESR and is unaffected by the level of Hb, but it is less helpful than the ESR or plasma viscosity in monitoring chronic inflammatory diseases. The measurement of CRP is easy and quick to perform using an immunoassay that can be automated. High-sensitivity assays have shown that increased levels may predict future cardiovascular disease (see p. 728).
Table 8.1 Normal values for peripheral blood
| Male | Female | |
|---|---|---|
Hb (g/L) | 135–175 | 115–160 |
PCV (haematocrit; L/L) | 0.4–0.54 | 0.37–0.47 |
RCC (1012/L) | 4.5–6.0 | 3.9–5.0 |
MCV (fL) | 80–96 | |
MCH (pg) | 27–32 | |
MCHC (g/L) | 320–360 | |
RDW (%) | 11–15 | |
WBC (109/L) | 4.0–11.0 | |
Platelets (109/L) | 150–400 | |
ESR (mm/h) | <20 | |
Reticulocytes | 0.5–2.5% (50–100 × 109/L) | |
ESR, erythrocyte sedimentation rate; Hb, haemoglobin; MCH, mean corpuscular haemoglobin; MCHC, mean corpuscular haemoglobin concentration; MCV, mean corpuscular volume of red cells; PCV, packed cell volume; RCC, red cell count; RDW, red blood cell distribution width; WBC, white blood count.
The red cell
Erythropoiesis
 Reticulocytes contain residual ribosomal RNA and are still able to synthesize Hb. They remain in the marrow for about 1–2 days and are released into the circulation, where they lose their RNA and become mature red cells (erythrocytes) after another 1–2 days. Mature red cells are non-nucleated biconcave discs.
Reticulocytes contain residual ribosomal RNA and are still able to synthesize Hb. They remain in the marrow for about 1–2 days and are released into the circulation, where they lose their RNA and become mature red cells (erythrocytes) after another 1–2 days. Mature red cells are non-nucleated biconcave discs.
 Nucleated red cells (normoblasts) are not normally present in peripheral blood, but are present if there is extramedullary haemopoiesis and in some marrow disorders (see leucoeryothroblastic anaemia, p. 413).
Nucleated red cells (normoblasts) are not normally present in peripheral blood, but are present if there is extramedullary haemopoiesis and in some marrow disorders (see leucoeryothroblastic anaemia, p. 413).

 Erythropoietin is a hormone which controls erythropoiesis. The gene for erythropoietin is on chromosome 7 and codes for a heavily glycosylated polypeptide of 165 amino acids. Erythropoietin has a molecular weight of 30 400 and is produced in the peritubular cells in the kidneys (90%) and in the liver (10%). Its production is regulated mainly by tissue oxygen tension. Production is increased if there is hypoxia from whatever cause, e.g. anaemia or cardiac or pulmonary disease. The erythropoietin gene is one of a number of genes that is regulated by the hypoxic sensor pathway. The 3′-flanking region of the erythropoietin gene has a hypoxic response element, which is necessary for the induction of transcription of the gene in hypoxic cells. Hypoxia-inducible factor 1 (HIF-1) is a transcription factor, which binds to the hypoxia response element and acts as a master regulator of several genes that are responsive to hypoxia. Erythropoietin stimulates an increase in the proportion of bone marrow precursor cells committed to erythropoiesis, and CFU-E are stimulated to proliferate and differentiate. Increased ‘inappropriate’ production of erythropoietin occurs in certain tumours such as renal cell carcinoma and other causes (see Table 8.15).
Erythropoietin is a hormone which controls erythropoiesis. The gene for erythropoietin is on chromosome 7 and codes for a heavily glycosylated polypeptide of 165 amino acids. Erythropoietin has a molecular weight of 30 400 and is produced in the peritubular cells in the kidneys (90%) and in the liver (10%). Its production is regulated mainly by tissue oxygen tension. Production is increased if there is hypoxia from whatever cause, e.g. anaemia or cardiac or pulmonary disease. The erythropoietin gene is one of a number of genes that is regulated by the hypoxic sensor pathway. The 3′-flanking region of the erythropoietin gene has a hypoxic response element, which is necessary for the induction of transcription of the gene in hypoxic cells. Hypoxia-inducible factor 1 (HIF-1) is a transcription factor, which binds to the hypoxia response element and acts as a master regulator of several genes that are responsive to hypoxia. Erythropoietin stimulates an increase in the proportion of bone marrow precursor cells committed to erythropoiesis, and CFU-E are stimulated to proliferate and differentiate. Increased ‘inappropriate’ production of erythropoietin occurs in certain tumours such as renal cell carcinoma and other causes (see Table 8.15).
Haemoglobin synthesis
Haemoglobin performs the main functions of red cells – carrying O2 to the tissues and returning CO2 from the tissues to the lungs. Each normal adult Hb molecule (HbA) has a molecular weight of 68 000 and consists of two α and two β globin polypeptide chains (α2β2). HbA comprises about 97% of the Hb in adults. Two other haemoglobin types, HbA2 (α2δ2) and HbF (α2γ2), are found in adults in small amounts (1.5–3.2% and <1%, respectively) (see p. 390).
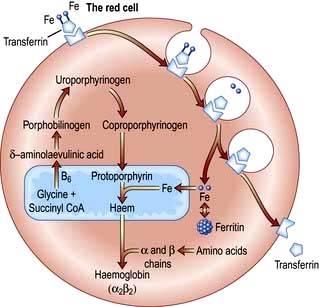
Haemoglobin synthesis occurs in the mitochondria of the developing red cell (Fig. 8.2). The major rate-limiting step is the conversion of glycine and succinic acid to δ-aminolaevulinic acid (ALA) by ALA synthase. Vitamin B6 is a coenzyme for this reaction, which is inhibited by haem and stimulated by erythropoietin. Two molecules of δ-ALA condense to form a pyrrole ring (porphobilinogen). These rings are then grouped in fours to produce protoporphyrins and with the addition of iron haem is formed. Haem is then inserted into the globin chains to form a haemoglobin molecule. The structure of Hb is shown in Figure 8.3.
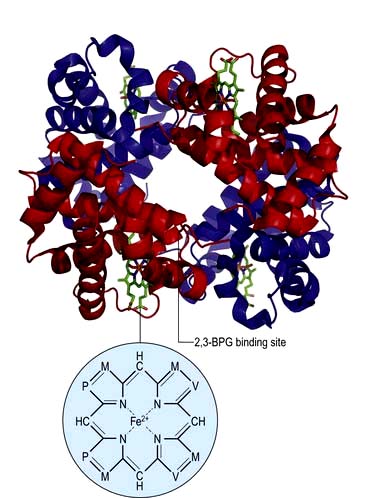
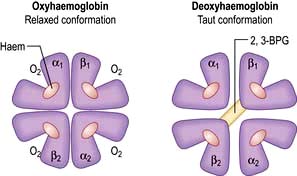
Haemoglobin function
In adult haemoglobin (Hb), a haem group is bound to each of the four globin chains; the haem group has a porphyrin ring with a ferrous atom which can reversibly bind one oxygen molecule. The haemoglobin molecule exists in two conformations, R and T. The T (taut) conformation of deoxyhaemoglobin is characterized by the globin units being held tightly together by electrostatic bonds (Fig. 8.4). These bonds are broken when oxygen binds to haemoglobin, resulting in the R (relaxed) conformation in which the remaining oxygen binding sites are more exposed and have a much higher affinity for oxygen than in the T conformation. The binding of one oxygen molecule to deoxyhaemoglobin increases the oxygen affinity of the remaining binding sites – this property is known as ‘cooperativity’ and is the reason for the sigmoid shape of the oxygen dissociation curve. Haemoglobin is, therefore, an example of an allosteric protein. The binding of oxygen can be influenced by secondary effectors – hydrogen ions, carbon dioxide and red-cell 2,3-bisphosphoglycerate (2,3-BPG). Hydrogen ions and carbon dioxide added to blood cause a reduction in the oxygen-binding affinity of haemoglobin (the Bohr effect). Oxygenation of haemoglobin reduces its affinity for carbon dioxide (the Haldane effect). These effects help the exchange of carbon dioxide and oxygen in the tissues.
A summary of normal red cell production and destruction is given in Figure 8.5.
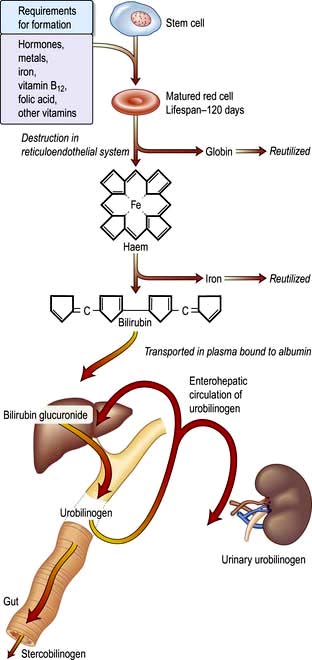
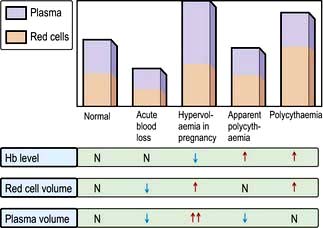
Anaemia
Anaemia is present when there is a decrease in Hb in the blood below the reference level for the age and sex of the individual (Table 8.1). Alterations in the Hb may occur as a result of changes in the plasma volume, as shown in Figure 8.6. A reduction in the plasma volume will lead to a spuriously high Hb – this is seen with dehydration and in the clinical condition of apparent polycythaemia (see p. 404). A raised plasma volume produces a spurious anaemia, even when combined with a small increase in red cell volume as occurs in pregnancy.
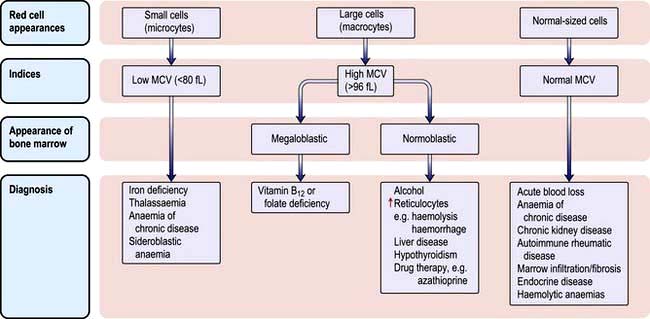
The various types of anaemia, classified by MCV, are shown in Figure 8.7. There are three major types:



Clinical features













Investigations
Peripheral blood
A low Hb should always be evaluated with:

 The white blood cell (WBC) count
The white blood cell (WBC) count

 The reticulocyte count (as this indicates marrow activity)
The reticulocyte count (as this indicates marrow activity)
 The blood film, as abnormal red cell morphology (see Fig. 8.9) may indicate the diagnosis. Where two populations of red cells are seen, the blood film is said to be dimorphic. This may, for example, be seen in patients with ‘double deficiencies’ (e.g. combined iron and folate deficiency in coeliac disease, or following treatment of anaemic patients with the appropriate haematinic).
The blood film, as abnormal red cell morphology (see Fig. 8.9) may indicate the diagnosis. Where two populations of red cells are seen, the blood film is said to be dimorphic. This may, for example, be seen in patients with ‘double deficiencies’ (e.g. combined iron and folate deficiency in coeliac disease, or following treatment of anaemic patients with the appropriate haematinic).
Bone marrow
Techniques for obtaining bone marrow are shown in Practical Box 8.1.
 Practical Box 8.1
Practical Box 8.1
Techniques for obtaining bone marrow
The technique should be explained to the patient and consent obtained.















Examination of the bone marrow is performed to further investigate abnormalities found in the peripheral blood (Practical Box 8.1). Aspiration provides a film which can be examined by microscopy for the morphology of the developing haemopoietic cells. The trephine provides a core of bone which is processed as a histological specimen and allows an overall view of the bone marrow architecture, cellularity and presence/absence of abnormal infiltrates.

 Type of erythropoiesis (e.g. normoblastic or megaloblastic)
Type of erythropoiesis (e.g. normoblastic or megaloblastic)
 Cellularity of the various cell lines
Cellularity of the various cell lines
 Infiltration of the marrow, i.e. presence of non-haematopoietic cells such as cancer cells
Infiltration of the marrow, i.e. presence of non-haematopoietic cells such as cancer cells

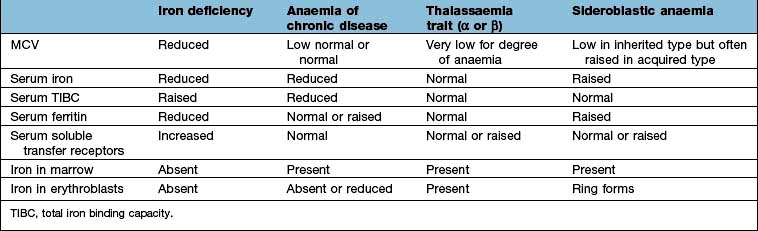
Microcytic anaemia
The other causes of a microcytic hypochromic anaemia are anaemia of chronic disease, sideroblastic anaemia and thalassaemia. In thalassaemia (see p. 390), there is a defect in globin synthesis, in contrast to the other three causes of microcytic anaemia where the defect is in the synthesis of haem.
Iron
Absorption
Factors influencing iron and haem iron absorption (Fig. 8.8) are shown in Table 8.2.
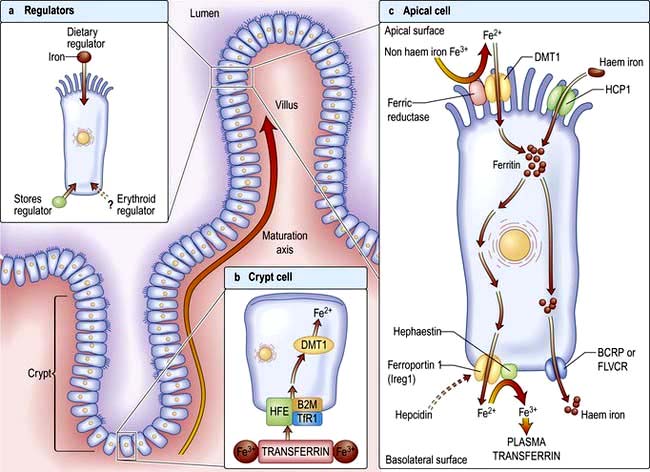
Table 8.2 Factors influencing iron absorption
Dietary haem iron is more rapidly absorbed than non-haem iron derived from vegetables and grain. Most haem is absorbed in the proximal intestine, with absorptive capacity decreasing distally. The intestinal haem transporter HCP1 (haem carrier protein 1) has been identified and found to be highly expressed in the duodenum. It is upregulated by hypoxia and iron deficiency. Some haem iron may be reabsorbed intact into circulation via the cell by two exporter proteins – BCRP (breast cancer resistant protein) and FLVCR (feline leukaemia virus subgroup C) (Fig. 8.8).
Once inside the mucosal cell, iron may be transferred across the cell to reach the plasma, or be stored as ferritin; the body’s iron status at the time the absorptive cell developed from the crypt cell is probably the crucial deciding factor. Iron stored as ferritin will be lost into the gut lumen when the mucosal cells are shed; this regulates iron balance. The mechanism of transport of iron across the basolateral surface of mucosal cells involves a transporter protein, ferroportin 1 (FPN 1) through its iron-responsive element (IRE). This transporter protein requires an accessory, multicopper protein, hephaestin (Fig. 8.8).
Transport in the blood
The normal serum iron level is about 13–32 µmol/L; there is a diurnal rhythm with higher levels in the morning. Iron is transported in the plasma bound to transferrin, a β-globulin that is synthesized in the liver. Each transferrin molecule binds two atoms of ferric iron and is normally one-third saturated. Most of the iron bound to transferrin comes from macrophages in the reticuloendothelial system and not from iron absorbed by the intestine. Transferrin-bound iron becomes attached by specific receptors to erythroblasts and reticulocytes in the marrow and the iron is removed (Fig. 8.2).


Iron deficiency




Clinical features
The symptoms of anaemia are described on page 375. The well known clinical features of iron deficiency listed below are generally only seen in cases of very longstanding iron deficiency:

 Spoon-shaped nails (koilonychia)
Spoon-shaped nails (koilonychia)
 Atrophy of the papillae of the tongue
Atrophy of the papillae of the tongue


 A syndrome of dysphagia and glossitis (Plummer–Vinson or Paterson–Brown–Kelly syndrome; see p. 243).
A syndrome of dysphagia and glossitis (Plummer–Vinson or Paterson–Brown–Kelly syndrome; see p. 243).
Investigations
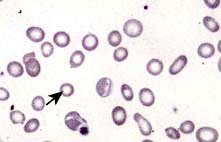
 Blood count and film. A characteristic blood film is shown in Figure 8.9. The red cells are microcytic (MCV <80 fL) and hypochromic (MCH (mean corpuscular haemoglobin) <27 pg). There is poikilocytosis (variation in shape) and anisocytosis (variation in size). Target cells are seen.
Blood count and film. A characteristic blood film is shown in Figure 8.9. The red cells are microcytic (MCV <80 fL) and hypochromic (MCH (mean corpuscular haemoglobin) <27 pg). There is poikilocytosis (variation in shape) and anisocytosis (variation in size). Target cells are seen.
 Serum iron and iron-binding capacity. The serum iron falls and the total iron-binding capacity (TIBC) rises in iron deficiency compared with normal. Iron deficiency is regularly present when the transferrin saturation (i.e. serum iron divided by TIBC) falls below 19% (Table 8.3).
Serum iron and iron-binding capacity. The serum iron falls and the total iron-binding capacity (TIBC) rises in iron deficiency compared with normal. Iron deficiency is regularly present when the transferrin saturation (i.e. serum iron divided by TIBC) falls below 19% (Table 8.3).
 Serum ferritin. The level of serum ferritin reflects the amount of stored iron. The normal values for serum ferritin are 30–300 µg/L (11.6–144 nmol/L) in males and 15–200 µg/L (5.8–96 nmol/L) in females. In simple iron deficiency, a low serum ferritin confirms the diagnosis. However, ferritin is an acute-phase reactant, and levels increase in the presence of inflammatory or malignant diseases. Very high levels of ferritin may be observed in hepatitis and in a rare disease, haemophagocytic lymphohistiocytosis (p. 80).
Serum ferritin. The level of serum ferritin reflects the amount of stored iron. The normal values for serum ferritin are 30–300 µg/L (11.6–144 nmol/L) in males and 15–200 µg/L (5.8–96 nmol/L) in females. In simple iron deficiency, a low serum ferritin confirms the diagnosis. However, ferritin is an acute-phase reactant, and levels increase in the presence of inflammatory or malignant diseases. Very high levels of ferritin may be observed in hepatitis and in a rare disease, haemophagocytic lymphohistiocytosis (p. 80).
 Serum soluble transferrin receptors. The number of transferrin receptors increases in iron deficiency. The results of this immunoassay compare well with results from bone marrow aspiration at estimating iron stores. This assay can help to distinguish between iron deficiency and anaemia of chronic disease (Table 8.3), and may avoid the need for bone marrow examination even in complex cases. It may sometimes be helpful in the investigation of complicated causes of anaemia.
Serum soluble transferrin receptors. The number of transferrin receptors increases in iron deficiency. The results of this immunoassay compare well with results from bone marrow aspiration at estimating iron stores. This assay can help to distinguish between iron deficiency and anaemia of chronic disease (Table 8.3), and may avoid the need for bone marrow examination even in complex cases. It may sometimes be helpful in the investigation of complicated causes of anaemia.
 Other investigations. These will be indicated by the clinical history and examination. Investigations of the gastrointestinal tract are often required to determine the cause of the iron deficiency (see p. 257).
Other investigations. These will be indicated by the clinical history and examination. Investigations of the gastrointestinal tract are often required to determine the cause of the iron deficiency (see p. 257).
Differential diagnosis
The presence of anaemia with microcytosis and hypochromia does not necessarily indicate iron deficiency. The most common other causes are thalassaemia, sideroblastic anaemia and anaemia of chronic disease, and in these disorders the iron stores are normal or increased. The differential diagnosis of microcytic anaemia is shown in Table 8.3.



Anaemia of chronic disease
The serum iron and the TIBC are low, and the serum ferritin is normal or raised because of the inflammatory process. The serum soluble transferrin receptor level is normal (Table 8.3). Stainable iron is present in the bone marrow, but iron is not seen in the developing erythroblasts. Patients do not respond to iron therapy, and treatment is, in general, that of the underlying disorder. Recombinant erythropoietin therapy is used in the anaemia of renal disease (see p. 623), and occasionally in inflammatory disease (rheumatoid arthritis, inflammatory bowel disease).
Sideroblastic anaemia
Sideroblastic anaemias are inherited or acquired disorders characterized by a refractory anaemia, a variable number of hypochromic cells in the peripheral blood, and excess iron and ring sideroblasts in the bone marrow. The presence of ring sideroblasts is the diagnostic feature of sideroblastic anaemia. There is accumulation of iron in the mitochondria of erythroblasts owing to disordered haem synthesis forming a ring of iron granules around the nucleus that can be seen with Perls’ reaction. The blood film is often dimorphic; ineffective haem synthesis is responsible for the microcytic hypochromic cells. Sideroblastic anaemias can be inherited as an X-linked disease transmitted by females. Acquired causes include myelodysplasia, myeloproliferative disorders, myeloid leukaemia, drugs (e.g. isoniazid), alcohol misuse and lead toxicity. It can also occur in other disorders such as rheumatoid arthritis, carcinomas, megaloblastic and haemolytic anaemias. A structural defect in δ-aminolaevulinic acid (ALA) synthase, the pyridoxine-dependent enzyme responsible for the first step in haem synthesis (Fig. 8.2), has been identified in one form of inherited sideroblastic anaemia. Primary acquired sideroblastic anaemia is one of the myelodysplastic syndromes (see p. 405) and this is the cause of the vast majority of cases of sideroblastic anaemia in adults. Lead toxicity is described in Chapter 17.
Normocytic anaemia
Normocytic, normochromic anaemia is seen in anaemia of chronic disease, in some endocrine disorders (e.g. hypopituitarism, hypothyroidism and hypoadrenalism) and in some haematological disorders (e.g. aplastic anaemia and some haemolytic anaemias) (Fig. 8.7). In addition, this type of anaemia is seen acutely following blood loss.
Macrocytic anaemias
Megaloblastic anaemia
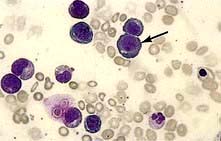
Megaloblastic anaemia is characterized by the presence in the bone marrow of erythroblasts with delayed nuclear maturation because of defective DNA synthesis (megaloblasts). Megaloblasts are large and have large immature nuclei. The nuclear chromatin is more finely dispersed than normal and has an open stippled appearance (Fig. 8.10). In addition, giant metamyelocytes are frequently seen in megaloblastic anaemia. These cells are about twice the size of normal cells and often have twisted nuclei. Megaloblastic changes occur in:
 Vitamin B12 deficiency or abnormal vitamin B12 metabolism
Vitamin B12 deficiency or abnormal vitamin B12 metabolism
 Folic acid deficiency or abnormal folate metabolism
Folic acid deficiency or abnormal folate metabolism


Haematological findings
 Anaemia may be present. The MCV is characteristically >96 fL unless there is a co-existing cause of microcytosis when there may be a dimorphic picture with a normal/low average MCV.
Anaemia may be present. The MCV is characteristically >96 fL unless there is a co-existing cause of microcytosis when there may be a dimorphic picture with a normal/low average MCV.
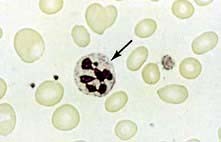
 The peripheral blood film shows oval macrocytes with hypersegmented polymorphs with six or more lobes in the nucleus (Fig. 8.11).
The peripheral blood film shows oval macrocytes with hypersegmented polymorphs with six or more lobes in the nucleus (Fig. 8.11).

Biochemical basis of megaloblastic anaemia
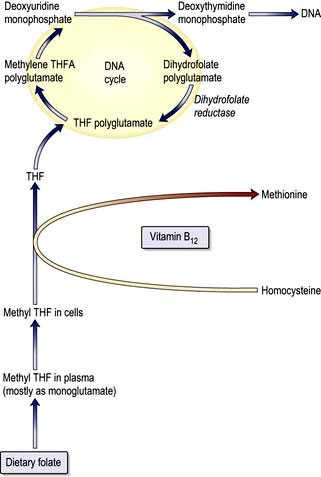
The key biochemical problem common to both vitamin B12 and folate deficiency is a block in DNA synthesis owing to an inability to methylate deoxyuridine monophosphate to deoxythymidine monophosphate, which is then used to build DNA (Fig. 8.12). The methyl group is supplied by the folate coenzyme, methylene tetrahydrofolate.
Vitamin B12
Structure and function
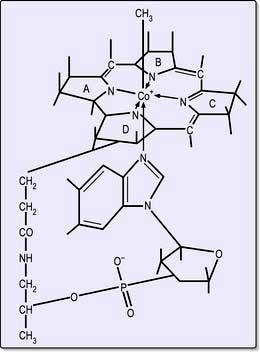
Cobalamins consist of a planar group with a central cobalt atom (corrin ring) and a nucleotide set at right-angles (Fig. 8.13). Vitamin B12 was first crystallized as cyanocobalamin, but the main natural cobalamins have deoxyadenosyl-, methyl- and hydroxocobalamin groups attached to the cobalt atom.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


