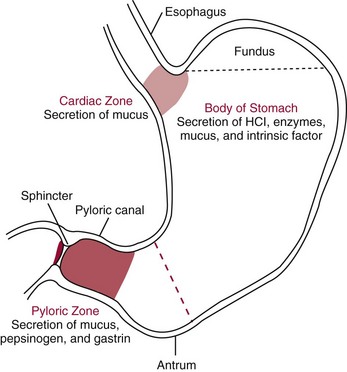
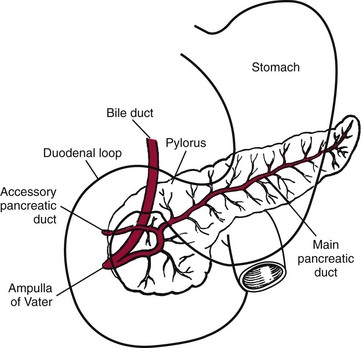
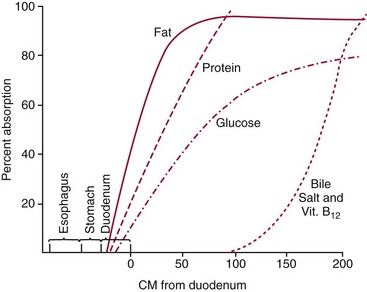
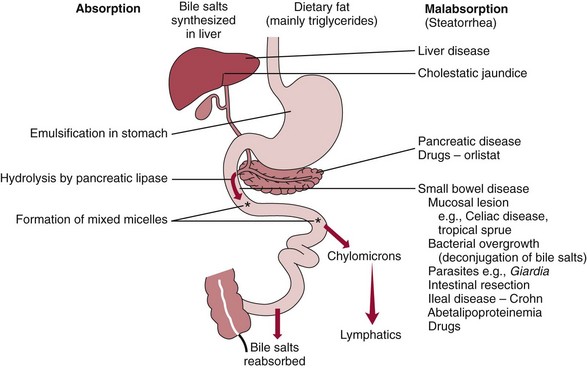
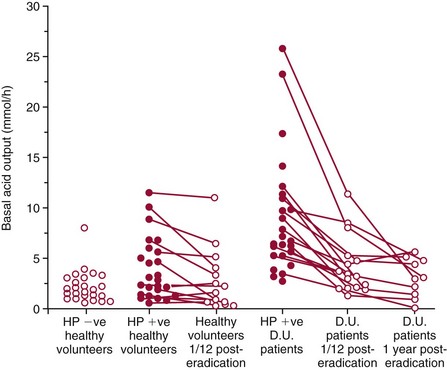
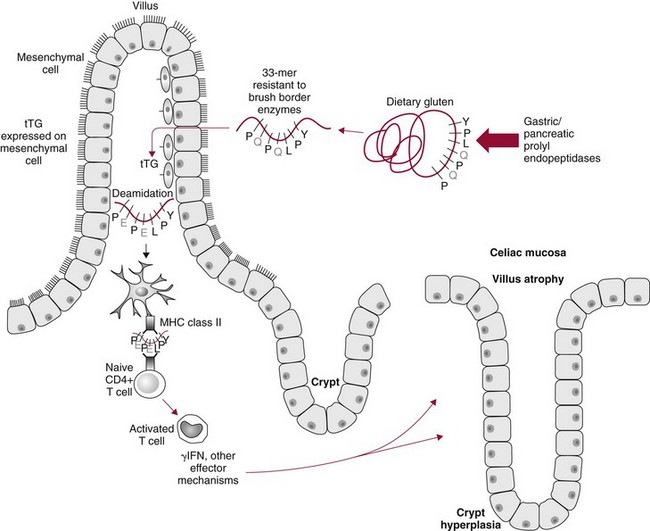
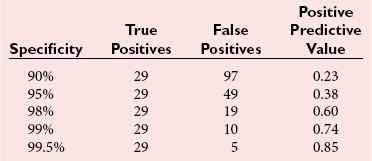
Chapter 51 The stomach consists of three major zones: the cardiac zone, the body, and the pyloric zone (Figure 51-1). The upper cardiac zone, which includes the fundus, contains mucus-secreting surface epithelial cells, which also secrete group II pepsinogens and several types of endocrine secreting cells. The body of the stomach contains cells or cell groups of many different types: (1) surface epithelial cells, which secrete mucus; (2) parietal (oxyntic) cells, which secrete hydrochloric acid and intrinsic factor; (3) the chief, zymogen, or peptic cells, which secrete group I and II pepsinogens; (4) enterochromaffin cells, which secrete serotonin; and (5) several types of endocrine secreting cells. The pyloric zone is subdivided into the antrum (which is approximately the distal third of the stomach), the pyloric canal, and the sphincter. The cells of the pyloric zone secrete mucus, group II pepsinogens, serotonin, gastrin, and several other hormones but no hydrochloric acid. The wall of the small intestine consists of four layers: mucous, submucous, muscular, and serous. The internal surface of the upper small intestine contains valvelike circular folds (valvulae conniventes or plicae circulares) that project 3 to 10 mm into the lumen of the intestine. Covering the entire mucous surface of the small intestine are very small (1 mm) finger-like projections (villi), giving it a “velvety” appearance. The luminal surface (brush border) of each epithelial cell consists of some 1700 microvilli projecting about 1 µm from the cell. The folds, villi, and microvilli together present an absorptive surface some 600 times greater than would be inferred from the length and diameter of this portion of the gut. The absorptive surface area of the small intestine is estimated to be about 250 m2,73 which is comparable with the area of a doubles tennis court. The pancreas is 12 to 15 cm in length and lies across the posterior wall of the abdominal cavity. The head is located in the duodenal curve; the body and tail are directed toward the left, extending to the spleen (Figure 51-2). Pancreatic digestive enzymes, in bicarbonate-rich juice, enter the duodenum through the ampulla of Vater and the sphincter of Oddi and mix with the food bolus as it passes through the small bowel. Digestion of ingested food takes place both within the lumen of the small intestine and at the mucosal (brush border) surface. Defects of digestion may occur at one or more stages in the process. The terms maldigestion and malabsorption refer to different functional abnormalities. Maldigestion is a dysfunction of the digestive process that may occur at various sites in the GI tract. For example, hypoacidity in the stomach will reduce peptic digestion of protein; hyperacidity of the duodenum (e.g., due to overproduction of gastrin by tumor in the Zollinger-Ellison syndrome) can inactivate pancreatic enzymes; loss of brush border enzymes in the small intestine, because of any of a variety of processes, can prevent oligosaccharides and disaccharides from being further hydrolyzed; pancreatic insufficiency will reduce intraluminal enzyme activity in the small gut, causing maldigestion of fats and proteins. By contrast, malabsorption is strictly a dysfunction of the absorptive process in the small gut due to loss of absorptive epithelial cells caused, for example, by gluten, inflammation, infection, surgical resection, and infiltration. Various transport defects also lead to malabsorption of specific substances (e.g., glucose-galactose malabsorption or zinc deficiency in the congenital disorder acrodermatitis enteropathica). In clinical practice, however, the term malabsorption is often used to encompass all aspects of impaired digestion and absorption. As Figure 51-3 shows, absorption of the different nutrients proceeds at different rates and at different sites within the small bowel. The brush border enzymes with disaccharidase and oligosaccharidase activity are listed in Table 51-1. The sucrase-isomaltase complex comprises most of the sucrase, isomaltase, and maltase (80%) activity of the small intestine. It hydrolyzes sucrose to its constituent monosaccharides, cleaves glucose from α-limit dextrins with 1,6 bonds, and hydrolyzes maltose. The activity of the complex is fourfold to fivefold greater in the jejunum than in the ileum. Changes in diet have a marked effect on the expression of the complex; starvation leads to a rapid decline in activity, which is rapidly restored on refeeding. All small intestinal saccharidases may decrease with infection or inflammation of the small bowel to the extent that carbohydrate malabsorption occurs, leading to diarrhea, flatulence, and weight loss. Paradoxically, diabetes mellitus causes a striking increase in sucrase-isomaltase activity; an increase is also observed in monosaccharide and amino acid transport. The lactase–phlorizin hydrolase complex is the only brush border enzyme able to hydrolyze lactose and therefore is essential for the survival of mammals early in life. TABLE 51-1 Brush Border Oligosaccharidases From Semenza G, Auricchio S, Mantei N. Small-intestinal disaccharidases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular bases of inherited disease, 8th edition. New York, NY: McGraw-Hill, 2001:1623-50. The recommended daily dietary fat intake in Europe and North America is 70 to 95 g. Less than 5 g/24 h is recovered in the feces, indicating the overall efficiency of the normal processes of fat digestion and absorption. Most dietary fat is in the form of long chain triacylglycerols (triglycerides). Pancreatic lipase is quantitatively the most important hydrolytic enzyme, but the contribution of gastric lipase to overall hydrolysis should not be underestimated. Gastric lipase is secreted by the gastric mucosa and normally accounts for up to 17.5% of fatty acids released from triglycerides following a meal.18 The enzyme has a wide pH optimum and is active in both the stomach and the duodenum. This nonpancreatic lipase may have a significant role in lipid digestion when pancreatic function is impaired and in the neonatal period before pancreatic lipase activity is fully developed. A lingual lipase is also present, secreted by the tongue, but is thought not to be of much significance normally in humans. Fats first are emulsified in the stomach by its churning action and are stabilized by interaction with luminal lecithin and protein fragments. The lingual and gastric lipases do not require bile salts or cofactors to function; they have a pH optimum of 3 to 6, and their action produces 1,2-diacylglycerols and fatty acids. These products further stabilize the surface of the triglyceride emulsion and in the duodenum promote the binding of pancreatic colipase. In addition, the liberated fatty acids stimulate release of CCK from the duodenal mucosa. From the lymphatics, chylomicrons enter the bloodstream via the thoracic duct and are distributed to the liver, adipose tissue, and other organs. Medium and short chain fatty acids (chain length <12 carbon atoms) in mixed triglycerides are preferentially split by lipases and pass into the aqueous phase, from which they are rapidly absorbed. Medium chain triglycerides can be absorbed without complete lipolysis and in the absence of bile. They do not require micellar solubilization and are transported from the intestinal epithelial cells predominantly via the hepatic portal vein. Figure 51-4 summarizes the processes involved in fat absorption and conditions that compromise the efficiency of one or more stages in the process of fat digestion and absorption leading to fat malabsorption.24 Although the presence of spiral-shaped organisms in the stomach has been acknowledged for many years, it was only in 1985 that the association was described between H. pylori (known then as Campylobacter pylori) and peptic ulcer disease.125 Most estimates suggest that the bacterium is present in the mucous layer of the stomach in half the population of the world. In Europe, 30 to 50% of adults, and in the United States, at least 20% of the adult population, are infected with the organism. In all cases, colonization with H. pylori causes a chronic inflammatory reaction in the gastric mucosa even when direct endoscopic observation of the mucosa appears normal. Carriers of the organism are at increased risk for gastric cancer (twofold to tenfold) and peptic ulcer (threefold to tenfold).12 Some of this increased risk is due to infection with strains of the organism that produce the cytotoxic CagA protein. About 90% of gastric cancer patients are infected with H. pylori, compared with 40 to 60% of age-matched controls.141,142 In a European study comparing the prevalence of H. pylori versus gastric cancer rates in 13 countries, a significant correlation was observed between infection rate and gastric cancer incidence and mortality.175 It is, however, important to remember that although a large proportion of gastric cancer can be attributed to infection with H. pylori, only in a minority of infected subjects will the inflammatory reaction progress to gastric cancer. Gastric cancer rates in Western countries have declined in recent decades, but the incidence remains high in less developed countries. At least 95% of patients with duodenal ulcer disease are infected with H. pylori, and eradication of the organism leads to healing of the ulcer and a reduction in relapse rates.147 Eradication of H. pylori is now the recommended treatment for patients with duodenal or gastric ulcer who are H. pylori–positive. Effective combined antibiotic and acid suppression regimens (using PPIs) are available with eradication rates of up to 90% after first-line treatment.64 However, increases in the prevalence of antibiotic resistance have led to the development of alternative treatment regimes to maintain high eradication rates.122A Infection with the organism, with or without duodenal ulceration, leads to increases in both basal and meal-stimulated serum gastrin concentrations, principally due to an increase in gastrin-17.134 Basal acid output is increased in H. pylori–positive subjects (Figure 51-5) and resolves completely after successful eradication of the organism. Hypergastrinemia is believed to be only one of the mechanisms leading to increased acid output. Studies using the neuropeptide gastrin-releasing peptide (GRP) suggest that impairment of inhibitory control mechanisms that regulate acid production may be responsible for the increased acid output associated with H. pylori infection.49 In addition to stimulating G cells of the antrum to release gastrin, which leads to acid secretion by parietal cells, GRP activates neuroendocrine pathways that inhibit gastric acid secretion—an effect that is mediated via peptides (including cholecystokinin and secretin) that stimulate release of the inhibitory peptide somatostatin from the gastric mucosa. H. pylori produces urease, and hydrolysis of endogenous urea to bicarbonate and ammonia may create a more hospitable microenvironment for survival of the organism in the stomach. Mammalian cells do not hydrolyze urea, and it was only in 1984 that “gastric urease” was associated with the presence of H. pylori.110 The ability of the organism to rapidly hydrolyze urea is the basis of urea breath tests and of direct urease tests on gastric biopsy samples. Numerous invasive and noninvasive diagnostic tests for H. pylori have been described (Box 51-1), and many have been reviewed.68 All tests in the “invasive” group necessitate oral gastroduodenoscopy with biopsy of the gastric mucosa; false-negative results may occur as the result of sampling errors, as colonization may be patchy. The antrum is the preferred biopsy site, but multiple biopsies from the anterior and posterior walls of the antrum and the body of the stomach are recommended for maximum diagnostic accuracy of this group of tests. False negatives may also occur when biopsy specimens are taken during treatment with PPIs or within 2 weeks of stopping PPI therapy. These drugs alter the intragastric distribution of H. pylori and suppress its activity.118 During PPI therapy, biopsies should be taken from the body and fundus to prevent false negatives. PPIs can also lead to falsely negative urea breath test results. If PPIs cannot be withheld for at least 2 weeks before a breath test, a negative result must be interpreted with caution. Histamine H2-receptor antagonists should be stopped at least 24 hours before a breath test. Antacids do not affect the test results. Tests for H. pylori are required for the diagnosis of infection and to ascertain, in some situations, whether eradication therapy has been successful. High sensitivity is required to ensure that positives are not missed; similarly, high specificity is essential to prevent inappropriate use of eradication therapy. The Maastricht III Consensus Guidelines123 recommend a “test and treat” strategy in adults with appropriate dyspeptic symptoms younger than 45 years using a breath test or a stool antigen test. The age limit may vary depending on local prevalence and the age distribution of gastric cancer (e.g., in the United Kingdom, testing and treatment are now an option in any patients with uncomplicated dyspepsia, although for those aged 55 years and older with unexplained and persistent recent-onset dyspepsia alone, referral should be made for urgent endoscopy).139 Successful eradication of H. pylori should be confirmed with the urea breath test or by a monoclonal antibody-based stool antigen test if urea breath tests are not available. Other national guidelines confirm the urea breath test as the preferred procedure, both for initial diagnosis and for confirmation of eradication.22A,86,139 Testing to confirm eradication should be done at least 4 weeks after completion of the course of treatment. Urea breath tests are simple to perform, and both sensitivity and specificity are greater than 95%. Urea labeled with 14C or 13C is given orally as a drink or a capsule to be swallowed with water; urease from gastric H. pylori rapidly hydrolyzes the ingested urea to produce labeled bicarbonate, which is absorbed into the blood and exhaled as 14CO2 or 13CO2. The principal advantages of the 13C-urea breath test over the 14C-urea breath test are the simplicity of breath collection and the avoidance of regulations and environmental issues related to the use and disposal of radioisotopes. In the 14C-urea breath test, CO2 in expired air is trapped in methanolic hyamine hydroxide as the patient exhales through a straw, which should be fitted with a one-way valve to ensure that the patient does not suck the trapping solution into the mouth. A color change of an indicator (thymol blue) in the solution shows that the required quantity of CO2 has been trapped. Scintillant is then added and 14CO2 measured. In the 13C-urea breath test, the patient blows through a straw into an empty 15 mL tube, which is then capped. 13CO2/12CO2 ratios are compared for basal and postdose samples using isotope ratio mass spectrometry or alternative infrared measurement methods.106,117 In the stool test, specific H. pylori antigens are detected in microtiter plates coated with polyclonal or monoclonal antibodies. Sensitivity and specificity are lower than for the 13C-urea breath test. Monoclonal antibody tests are recommended for posteradication testing if the urea breath test is not available.123 Although still widely available, serologic tests are recommended only in specific situations (e.g., when PPI therapy cannot be withheld, when a patient with a bleeding ulcer is investigated).123 The systemic antibody response is variable, leading to equivocal results in some subjects; in subjects older than 50 years, diagnostic accuracy is unsatisfactory. Serology cannot be used to confirm eradication because of the slow decline in antibody levels after treatment. Laboratory-based enzyme-linked immunosorbent assays (ELISAs) and point-of-care tests are available to measure specific immunoglobulin (Ig)G antibodies in serum or whole blood samples. In younger subjects, laboratory-based tests generally perform well, although some have sensitivity and/or specificity less than 95%. Office-based serology testing has inadequate sensitivity and specificity and currently is not recommended.123 Calculations based on reported diagnostic accuracy data show that when these tests are used for diagnosis, as many as 28% of those receiving eradication therapy are being treated as a result of false-positive test results.23 1. Measure and transfer a convenient volume of gastric juice (e.g., 5.0 to 10.0 mL) into a clean titration vessel. If the gastric juice contains food particles or mucus, centrifuge the sample or filter it through gauze. 2. Determine the pH of the gastric specimen with a pH meter. If the pH is above 3.5, no free acid is present. Such a specimen need not be titrated. 3. Titrate the sample with NaOH, 0.10 mol/L, to a pH of 3.5, using a pH meter. If 5 mL of gastric specimen is titrated, the calculation becomes as follows: Gastric juice pH: if the pH is greater than 2.5, then it is unlikely that the raised gastrin is caused by Zollinger-Ellison syndrome. BAO: Normal: male 0 to 10.5 mmol/h; female: 0 to 5.6 mmol/h Zollinger-Ellison syndrome: 15 to 100 mmol/h, or greater than 5 mmol/h if acid-reducing surgery was performed previously. Celiac disease is sometimes called nontropical sprue, celiac sprue, or gluten-sensitive enteropathy. Recent evidence indicates that oats do not lead to an immune-mediated response nor to mucosal damage in subjects with celiac disease.90 Addition of oats to the list of permitted cereals increases choices and would be welcomed by most subjects with celiac disease. However, if oats are to be introduced into the diet, they must be obtained from a reliable source to ensure no contamination from wheat, barley, or rye proteins at any stage in the process from harvesting to packaging. The identification in 1997 of small bowel tissue transglutaminase (tTG)-2 as the autoantigen of celiac disease36 was a major step forward in understanding the pathogenesis of this disorder. The tissue transglutaminases are a family of calcium-dependent cytoplasmic enzymes that are released from cells during wounding. Although their physiologic role is incompletely understood, they are able to catalyze the cross-linking of proteins, leading to stabilization of the wound area. Expression of the enzyme is increased during apoptosis and in active celiac disease. It selectively cross-links or deamidates protein-bound glutamine residues. Deamidation of gliadin peptides by the enzyme enhances their binding to HLA DQ2/DQ8 and increases recognition of these peptides by gut-derived T cells from subjects with celiac disease.163 The pathogenesis of the disease is therefore believed to involve an interaction between tissue transglutaminase and gliadin peptides in genetically susceptible individuals. The toxic cereal proteins lead to intestinal epithelial damage, releasing tissue transglutaminase. Cross-linking by the enzyme produces gliadin-gliadin or gliadin-enzyme complexes, unmasking new antigenic epitopes that bind to HLA DQ2 molecules on the antigen-presenting cells, producing an immune response by gut-derived T cells. The characteristic enteropathy is then induced by the release of interferon-γ and other proinflammatory cytokines, as outlined in Figure 51-6. A 33-mer peptide of gluten appears to be the primary initiator of the inflammatory response.156 It is resistant to breakdown by all gastric, pancreatic, and intestinal brush border membrane proteases, thus allowing it to reach the small intestine intact. After deamidation by tissue transglutaminase, it is a potent inducer of gut-derived human T-cell lines from patients with celiac disease. Homologs of the peptide are found in food grains that are toxic to patients with celiac disease, but are absent from nontoxic food grains. The peptide could be detoxified by exposure to a bacterial prolyl endopeptidase, suggesting a therapeutic strategy for celiac disease.156 Increased intestinal permeability in untreated celiac disease that is reversible on withdrawal of gluten from the diet has been recognized since the early 1980s.74 Evidence suggests that this may be mediated by increased expression of zonulin,56 a protein that opens small intestinal tight junctions, or by decreased expression of intercellular epithelial cell adhesion molecules, such as Z0-1, catenin, and cadherin.144 The zonulin pathway is now thought to play a significant role in the entry of allergens into the cell and hence in the autoimmune response.54A Celiac disease is a common chronic disorder in Caucasian populations, with a prevalence of about 1%.122,186 It also occurs in northern Indian and North African populations. It is rare among Chinese, Japanese, and African-Caribbean people. It was previously considered to be a rare disorder in North America, but recent serologic and histologic evidence shows that the disease has been underdiagnosed, and that its prevalence is comparable, as might be expected, with that found in Europe.55,77 A wide spectrum has been noted in the clinical presentation of celiac disease, with most diagnoses made in adult life. Classical celiac disease, presenting in infancy up to the age of 2 years, with failure to thrive, abdominal distention, and diarrhea, is now an uncommon presentation. The spectrum of presenting symptoms in adults has changed over the past 20 years, and frank malabsorption is now uncommon.87,185 Most adults present now with nonspecific symptoms; mild iron deficiency is common. A strong association with other autoimmune disease, especially with type 1 diabetes mellitus and autoimmune thyroid disease, has been reported. In type 1 diabetes, the prevalence of celiac disease is about 5%, and serologic screening to detect these cases has been advocated.84 The initial presentation may be seen by a wide range of clinical specialties, as shown in Table 51-2. To make the diagnosis, there must be a high index of suspicion, along with awareness of the wide range of nonspecific symptoms and easy availability of serologic tests to select those patients in whom endoscopy is indicated to confirm the diagnosis. TABLE 51-2 Table 51-3 compares the sensitivity and specificity of the four IgA class antibodies commonly used. Both antireticulin (ARA) and endomysial (EMA) antibodies are measured by indirect immunofluorescence, ARA on rat kidney sections and EMA on monkey esophagus or human umbilical cord sections. The presence of ARA indicates the need to measure antibodies with higher specificity before small bowel histology is recommended for confirmation of the diagnosis. ARA and EMA are tissue type–dependent methods that detect autoantibodies to tissue transglutaminase-2.107 TABLE 51-3 Comparison of Serologic Tests for Celiac Disease ELISA, Enzyme-linked immunosorbent assay; Ig, immunoglobulin. Lack of standardization of assays for IgA-antigliadin antibodies (AGA) contributes to the variable diagnostic accuracy of this marker,81 but the sensitivity and specificity of AGA are poor, and use of this test should be abandoned. The sensitivity and specificity of current deamidated gliadin peptide antibody tests offer no advantages over tissue transglutaminase antibody (TGA) and may give significantly more false positives in subjects with liver disease.80,136 The sensitivity of EMA in some reports is compromised by selection bias,115 but most larger series in which patients have not been selected for a biopsy on the basis of positive serology indicate that the true sensitivity for EMA is between 90% and 95% (i.e., 5 to 10% of subjects with celiac disease have a negative EMA at diagnosis). When carried out correctly, the assay has very high specificity (>99%); laboratories should monitor their performance, as small reductions in specificity will lead to a significant increase in the numbers of patients subjected unnecessarily to a small bowel biopsy. Table 51-4 shows the effects of test specificity on the numbers of true and false positives per 1000 subjects tested, and on the positive predictive value at disease prevalence (in the population tested) of 3%, assuming a sensitivity of 95%. Many commercial kits are now available to measure IgA antibodies against tissue transglutaminase [“transglutaminase antibody” (TGA)] using human recombinant tissue transglutaminase or purified human enzyme as antigen. Their role in the investigation of celiac disease has been reviewed.80 Lack of standardization and differences in recombinant technology (e.g., the use of eukaryotic or prokaryotic organisms to produce tTG) can lead to variable performance.11,126 Specificity should be evaluated using a large series of samples representative of those routinely tested, and procedures should be selected on the basis of high specificity (minimum 99%) and high sensitivity (>90%). The use of TGA has advantages over EMA. Unlike EMA, which may be subject to observer bias, TGA is a quantitative procedure that does not require the use of primate tissue. It can be automated and therefore is appropriate for larger numbers of samples. This test has replaced EMA as the antibody of choice for performing serologic testing and for assessing dietary compliance of subjects on a gluten-free diet.14,79 As with all tests, not all kits perform to the same high standards, and laboratories should ensure that they participate in external quality assessment programs and select a well-validated method.45,80 In view of the growing public interest in celiac disorder, further standardization of these kits is urgently needed. For diagnosis, current guidelines require a jejunal biopsy with the characteristic villous atrophy, increased intraepithelial lymphocytes, and hyperplasia of the crypts.124 Wider use of serology has led to the recognition of more cases with minimal histologic changes and to use of the terms potential and latent celiac disease.83 Patients with positive EMA or TGA are described as having potential celiac disease when the jejunal biopsy has normal villi but shows subtle changes, such as increased numbers of intraepithelial lymphocytes. It is thought that in time these patients will develop a flat mucosa. The term latent celiac disease is used to describe patients who have at some time had positive antibodies and a flat mucosa that recovers on a gluten-free diet, but have also been found to have a normal mucosa while on a normal diet. Subjects with selective IgA deficiency (IgA <0.05 g/L, incidence about 1 : 600) are at greater risk of celiac disease. It is therefore important to identify these individuals rather than risk excluding the diagnosis on the basis of a negative test for IgA class antibodies.22 When EMA is used, total serum IgA should be measured on all samples submitted for celiac serology to identify those patients with IgA deficiency. A TGA method should be used that can distinguish those with a “normal” concentration from the small proportion of subjects with very low concentrations, in whom total IgA measurement is indicated to confirm or exclude IgA deficiency.57 When IgA deficiency is identified, IgG antibodies (IgG-EMA, IgG-TGA, and IgG anti-deamidated gliadin peptide) should be used as serologic screening tests.21 In view of the increased risk of the disorder in IgA deficiency, a small bowel biopsy should be considered in all IgA-deficient subjects with symptoms of celiac disease.22 Strict adherence to a gluten-free diet leads to mucosal recovery in celiac disease and reduces the risk of bowel malignancy. TGA can be used as a marker for monitoring dietary compliance, in addition to its diagnostic role.14 Failure of symptoms to respond to a prescribed gluten-free diet may indicate (1) nonadherence to the diet, (2) other coexisting conditions (such as small bowel bacterial overgrowth, lactose intolerance, and microscopic colitis), or (3) the presence of refractory celiac disease. Refractory celiac disease is characterized by persistent villous atrophy with an increase in intraepithelial lymphocytes in the small bowel while the patient is on a long-term gluten-free diet. In both responsive and refractory celiac disease, celiac antibodies typically are decreased with dietary therapy and remain within reference intervals unless individuals are re-exposed to gluten. Two types of refractory celiac disease may occur and are differentiated by the types of T-cell populations found in the intestinal mucosa—polyclonal in type I disease and clonal in type II disease. Differentiation of the two types is accomplished by immunohistochemical, flow cytometric, or T-cell receptor γ gene rearrangement analysis of intestinal mucosal T cells.129A Type II celiac disease has an unfavorable prognosis and typically is viewed as a precursor to enteropathy-associated T-cell lymphoma (EATL). Adult-type hypolactasia is the single most common absorptive defect in adults, with an incidence of 5 to 90% depending on the racial group, as shown in Table 51-5. TABLE 51-5 Prevalence of Hypolactasia in Adults In most humans (and in all other mammals), expression of the enzyme decreases during childhood, and by adulthood lactase levels are 10% or less of those seen in infancy. If symptoms of flatulence, abdominal discomfort, bloating, or diarrhea occur after consumption of one or two glasses of milk or of a large portion of ice cream or yogurt, lactose intolerance should be suspected. Suspicion would be increased in a subject from an ethnic group with a high incidence of lactose intolerance (see Table 51-5). Many methods have been proposed for detecting lactase deficiency (Box 51-2). Disaccharidase activities can be measured in homogenates of an intestinal biopsy.32 These assays are rarely required for routine diagnostic purposes, but when necessary (e.g., in investigations in infancy), they must be carried out by laboratories with expertise in these tests. Breath hydrogen is now the preferred test for diagnosing lactase deficiency. The use of hydrogen breath tests in disorders of carbohydrate absorption and in bacterial overgrowth has been reviewed.159 Oral tolerance tests, measuring the increase in plasma glucose or galactose following ingestion of lactose, have been used to diagnose lactase deficiency. The usual dose of lactose is 50 g in 200 mL water; lower doses should be used in children (2 g/kg, up to a maximum of 50 g). Multiple blood samples are collected over a 2 hour period and the peak increment in glucose (or galactose) noted. To exclude lactase deficiency, the increase above baseline for capillary plasma glucose should be greater than 1.1 mmol/L138 (20 mg/dL) or greater than 1.4 mmol/L (25 mg/dL) when venous plasma is used.20 In a survey of laboratory practice in the United Kingdom, widely varying cutoffs were found to be in use (1.0 to 2.7 mmol/L) even with the same lactose dose.44 The requirement for multiple blood samples and lack of procedural standardization suggest that these tests should be abandoned in favor of noninvasive breath hydrogen testing. Hydrogen is not an end product of mammalian metabolism, and breath hydrogen is derived from bacterial metabolism in the intestinal tract.114 Following lactose ingestion, the disaccharide normally will be split into its constituent monosaccharides and absorbed. With lactase deficiency, unabsorbed disaccharide will pass into the large bowel, and bacterial metabolism will produce hydrogen that is absorbed into the systemic circulation and exhaled in the breath. Breath hydrogen can be measured in end-expiratory breath with the use of laboratory or hand-held direct-reading electrochemical hydrogen monitors. Patient Preparation: Appropriate patient preparation is essential to ensure stable baseline breath hydrogen levels (Box 51-3). Avoidance of wheat-based foods and fiber for 12 hours before the test minimizes the availability of substrates for bacterial metabolism in the large bowel. Fasting breath hydrogen is typically less than 5 ppm (5 µL/L), and concentrations greater than 20 ppm (20 µL/L) may be an indication of malabsorption or bacterial overgrowth.143 Oral hygiene before ingestion of the substrate in hydrogen breath tests minimizes the production of hydrogen by oral bacteria. Brushing of teeth or use of an antibacterial mouthwash (e.g., 1% chlorhexidine) is recommended.128 Mouthwash containing alcohol should not be used, because this may interfere with the measurement of hydrogen. Cigarette smoke contains high hydrogen levels; smoking therefore is not permitted immediately before or during the test. Box 51-3 Protocol for Lactose Tolerance Test With Measurement of Breath Hydrogen Meal before 0700 (restriction on wheat and fiber), then fasting until test completed Brush teeth (night and morning) or use mouthwash Measure end-expiratory fasting breath H2 Give lactose solution (50 g in 180 mL water for adults) Rinse mouth with further 20 mL water and swallow Measure breath H2 at 15, 30, 60, 90, and 120 minutes Test can be stopped if earlier rise of greater than 20 ppm above fasting level
Gastric, Pancreatic, and Intestinal Function
Introduction to Anatomy and Physiology of the Gastrointestinal Tract176
Anatomy
Stomach152
Small Intestine
Pancreas
Processes of Digestion And Absorption52
Digestion and Absorption of Carbohydrates
Enzyme or Complex
Principal Substrate(s)
Products
β-Glycosidase Complex
Lactase (EC 3.2.1.23)
Lactose
Glucose + galactose
Glycosylceramidase (EC 3.2.1.45-46) [also called phlorizin hydrolase (EC 3.2.1.62)]
Galactosyl and glucosyl-β-ceramides
Galactose/glucose + ceramides
Sucrase-Isomaltase Complex
Sucrase-(maltase) (EC 3.2.1.48)
Maltose/sucrose
Glucose/fructose + glucose
Isomaltase-(maltase) (EC 3.2.1.10)
1,6-α-linkages in isomaltose and α-dextrins
Maltose
Glucose
Glucose
Trehalase (EC 3.2.1.28)
Trehalose
Glucose
Glucoamylase Complex (EC 3.2.1.20)
Glucoamylase-(maltase)-1 and glucoamylase-(maltase)-2 (have similar substrate specificities)
1,4-α-linkages at nonreducing ends of amylose, amylopectin, glycogen, and straight chain 1,4-α-glucopyranosyl oligomers, including maltose
Glucose
Digestion and Absorption of Lipids
Stomach: Diseases and Laboratory Investigations
Peptic Ulcer Disease and Helicobacter pylori64,123,158,180
Diagnostic Tests for H. pylori
Determination of Basal Acid Output35,91
Determination of Free Hydrochloric Acid in Gastric Juice
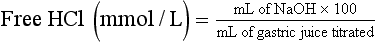
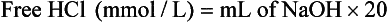
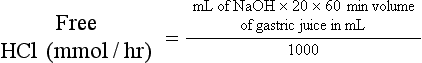
Interpretation
Intestinal Disorders and Their Laboratory Investigation
Celiac Disease (Celiac Sprue, Gluten-Sensitive Enteropathy)53,54,124,162,181A
Pathophysiology of Celiac Disease
Clinical Considerations
Clinical Specialty
Symptoms/Manifestations
General medicine
Tired all the time
Gastroenterology
Diarrhea, flatulence, weight loss
Hematology
Anemia
Obstetrics/gynecology
Infertility
Orthopedics
Fracture, osteopenia
Dermatology
Dermatitis herpetiformis, hyperkeratosis
Neurology
Peripheral neuropathy
Rheumatology
Arthropathy
Endocrinology
Short stature, thyroid disease
Diabetes
Diarrhea, anemia
Tests for Celiac Disease

Disaccharidase Deficiencies155,171
Racial Group
Prevalence, %
Chinese
>90
American blacks
54-81
Asians
60-90
Greeks
60-78
Northern Europeans
5-30
Adult-type Hypolactasia (“Acquired Lactase Deficiency”)
Diagnostic Tests for Lactase Deficiency
Oral Lactose Tolerance Tests
Hydrogen Breath Tests
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree