Flaviviruses
Theodore C. Pierson
Michael S. Diamond
Flaviviruses acquired their name from the jaundice associated with the liver dysfunction caused by yellow fever virus (YFV) infections. YFV played an important historical role in defining the nature of viruses in general. Seminal studies by Walter Reed and colleagues780 demonstrated that the etiology of yellow fever was a filterable agent that could be transmitted through the bite of a mosquito, confirming the postulates of Carlos Finlay. YFV was the first flavivirus isolated (in 1927) and the first to be propagated in vitro.790,820 These advances led remarkably rapidly to the development of an effective YFV vaccine that remains in use today.569 Experiments with the louping ill virus (LIV) in 1931 established that ticks also were capable of transmitting viruses associated with human disease.119 The discovery that antisera raised against some, but not all, viruses that caused similar diseases (e.g., encephalitis) cross-reacted with heterologous viruses provided a method to investigate the relatedness of flaviviruses.126,127,128,876 This was refined further with the development of a standardized hemagglutination inhibition test that allowed classification of 10 different flaviviruses and distinguished them from alphaviruses.127 These two groups of viruses were referred to thereafter as group A and B arboviruses, respectively. The first full-length flavivirus genome (YFV) were sequenced in 1985 by Charles Rice and colleagues.680 Subsequent advances in the molecular genetics of flaviviruses have increased our understanding of the relationships between viruses that was originally revealed by serology (described below). Seventy-three viruses of the Flavivirus genus (classified as 53 distinct species) have since been defined (http://www.ICTVonline.org/index.asp).
Flavivirus Diversity, Evolution, and Distribution
Molecular Phylogeny
Phylogenic relationships among members of the flavivirus genus have been established through the analysis of individual genes, and, more recently, the entire open reading frame of the genome.73,160,248,274,275,402,435,910 Analysis of the phylogenic tree of flaviviruses with respect to key features of the biology and ecology of these viruses has proven insightful.248,271 Three groups of viruses are defined based on their mode of transmission: tick-borne flaviviruses (TBFVs), mosquito-borne flaviviruses viruses (MBFVs), and those flaviviruses with no known vector (NKV). The earliest divergence from a monophyletic origin separates flaviviruses based on their mode of transmission.435,528 One lineage arising from the earliest branch of the phylogenic tree contains viruses transmitted by ticks and two groups of NKV viruses (Fig. 26.1). The second includes the mosquito-borne viruses and the Entebbe bat virus (ENTV) group of NKV viruses.402 The MBFVs are grouped further as a function of their association with mosquitoes of the Aedes and Culex genera. Viruses in these two clades cause hemorrhagic disease and encephalitis in humans and livestock, respectively. Although many of the viruses in the Culex clade infect avian hosts, Aedes viruses generally do not. Conversely, Culex viruses are not maintained in nature in infection cycles involving primates.
The tick-borne flaviviruses include 12 species divided into three groups274,402 (Fig. 26.1). The largest group of TBFVs is associated with mammalian hosts (typically rodents), and includes viruses that cause encephalitis (e.g., tick-borne encephalitis viruses [TBEVs]) and hemorrhagic fever (Omsk hemorrhagic fever virus [OHFV] and Kyasanur Forest disease virus [KFDV]) in humans. In addition, three species of mammalian TBFVs have not been associated with disease (Royal Farm virus [RFV], Karshi virus [KSIV], and Gadgets Gully virus [GGYV]). Viruses of the TBEV serocomplex are thought to represent a continuous evolutionary cline (a genetic gradient) that originated in Africa and moved from east to west across the Northern Hemisphere; the genetic distance between viruses in the mammalian tick-borne group correlates with increases in geographic distance.274,910 This is reflected by the asymmetric branching pattern of the phylogenic tree of the mammalian viruses911 (Fig. 26.1). The evolution of RFV, KSIV, and GGYV is not associated with this TBEV serocomplex cline, nor understood. This second group of TBFVs replicates within seabirds and ornithophilic ticks but does not cause disease in humans. These viruses have a broad geographic range that presumably reflects the migratory patterns of their avian hosts.271,274 The sole member of the final group of TBFVs is the Kadam virus (KADV). KADV is found in Africa and is typically associated with livestock.271 An understanding of the relationship between KADV and other members of the TBFVs has evolved. Although these viruses have been assigned to both the mammalian or sea bird groups of flaviviruses, a more recent analysis of the complete coding sequence of TBFVs places this virus in its own group, which is supported by unique features of its envelope protein and the fact that it encodes a polyprotein that is smaller than the rest of the TBFVs.274
MBFVs diverged early into two lineages; viruses within each of these lineages are subdivided based on their association with mosquitoes of the Aedes genus or Culex genus275,402 (Fig. 26.1). Viruses of the Aedes clade are a paraphyletic group thought to predate and give rise to the Culex viruses.271 One branch of the MBFV portion of the phylogenic tree includes viruses of the YFV group, the recently proposed Edge Hill virus (EHV) group, and two NKV viruses, discussed below. The YFV group includes Wesselsbron virus (WESSV), Sepik virus (SEPV), and YFV. WESSV is a veterinary pathogen transmitted by Aedes mosquitoes that causes a nonfatal febrile illness in humans. Very little is known about the clinical significance and vector biology of SEPV infection. Both SEPV and WESSV are found in Africa and Asia.275 The seven viruses of the EHV group are transmitted predominantly by Aedes mosquitoes, are found primarily in Africa (except for EHV, which is present in Australia), and share the unique property of encoding five (instead of six) disulfide bridges in the envelope glycoprotein. Human cases have been associated only with Banzi virus infection (BANV).275 The second branch of the MBFV phylogenic tree contains dengue viruses (DENVs), which are transmitted by Aedes mosquitoes, a large group of viruses vectored by Culex mosquitoes (e.g., Japanese encephalitis virus [JEV], West Nile virus [WNV]), and a group of Aedes-vectored viruses closes related to the Culex flaviviruses (e.g., Spondweni virus, SPOV).402 The diversity of viruses in the JEV serocomplex and DENV is described in detail below.
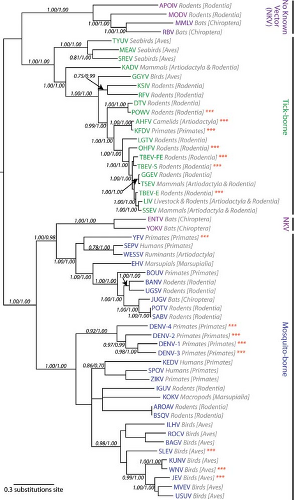 Figure 26.1. Phylogenetic tree of viruses in the genus Flavivirus reveals the evolutionary relationships among viruses transmitted by different vectors. A maximum-likelihood tree was generated using the complete polyprotein sequence of the indicated flaviviruses as detailed by Kitchen and colleagues.402 The viral taxa are abbreviated and colored according to their mode of transmission. The host reservoir for each virus is indicated in gray. Viruses that frequently cause disease in humans are indicated with red asterisks. APOIV, Apoi virus; MODV, Modoc virus; MMLV, Montana myotis leukoencephalitis virus; RBV, Rio Bravo virus; TYUV, Tyuleniy virus; MEAV, Meaban virus; SREV, Saumaurez Reef virus; KADV, Kadam virus; GGYV, Gadgets Gully virus; KSIV, Karshi virus; RFV, Royal Farm virus; DTV, Deer tick virus; POWV, Powassan virus; AHFV, Alkhurma hemorrhagic fever virus; KFDV, Kyasanur Forest disease virus; LGTV, Langat virus (LGTV); OHFV, Omsk hemorrhagic fever virus; TBEV-FE, Tick-borne encephalitis virus-far eastern subtype; TBEV-S, Tick-borne encephalitis virus-Siberian subtype; GGEV, Greek goat encephalomyelitis virus; TSEV, Turkish sheep encephalitis virus; TBEV-E, Tick-borne encephalitis virus-European subtype; LIV, Louping ill virus; SSEV, Spanish sheep encephalomyelitis virus; ENTV, Entebbe bat virus; YOKV, Yokose virus; YFV, Yellow fever virus; SEPV, Sepik virus; WESSV, Wesselsbron virus; EHV, Edge Hill virus; BOUV, Bouboui virus; BANV, Banzi virus; UGSV, Uganda S virus; JUGV, Jugra virus; POTV, Potiskum virus; SABV, Saboya virus; DENV-4, Dengue virus serotype 4; DENV-2, Dengue virus serotype 2; DENV-1, Dengue virus serotype 1; DENV-3, Dengue virus serotype 3; KEDV, Kedougu virus; SPOV, Spondweni virus; ZIKV, Zika virus; IGUV, Iguape virus; KOKV, Kokobera virus; AROAV, Aroa virus; BSQV, Bussuquara virus; ILHV, Ilheus virus; ROCV, Rocio virus; BAGV, Bagaza virus; SLEV, St. Louis encephalitis virus; KUNV, Kunjin virus; WNV, West Nile virus; JEV, Japanese encephalitis virus; MVEV, Murray Valley encephalitis virus; and USUV, Usutu virus. The tree was kindly provided by Dr. Edward Holmes and modified with permission. |
By comparison to the vector-borne flaviviruses, relatively little is known about the NKV viruses. These viruses are most commonly found in rodents or bats, in which they do not appear to cause disease or a high viremia. NKV viruses of rodents are found only in the New World, whereas those capable of infecting bats are found in both the Old and New Worlds.270 Although the majority of NKV viruses (12 species) diverged with the TBFVs, two species of bat-associated viruses appear to have evolved from a lineage transmitted by mosquitoes and then lost this trait secondarily (Yokose virus [YOKV] and ENTV)270,434,806 (Fig. 26.1). These viruses are most similar to the Aedes species–vectored viruses of the YFV group. Of interest, studies with chimeric YFV and DENV engineered to express the envelope genes of Modoc virus (MODV) suggest that the host-restriction of NKV viruses may be at a postentry step of the virus life cycle.138
Evolution
Many flaviviruses are transmitted by insect vectors, which is a unique feature of the genus not shared by the pestiviruses and hepaciviruses of the Flaviviridae family. One hypothesis is that flaviviruses evolved from an ancestral virus that was not vectored. Phylogenic analysis of NS5 sequences supports this notion271,435; these studies indicate that the majority of NKV viruses diverged from a lineage that gave rise to vector-borne viruses early in the evolution of flaviviruses. In this context, MBFVs arose from the TBFV lineage. An alternative hypothesis, supported by the analysis of NS3 and complete genome sequences, is that MBFVs diverged first and then gave rise to TBFVs and the majority of NVK viruses73,275,274,402 (Fig. 26.1). Flaviviruses likely originated in the Old World during the last 10,000 years since the last ice age.160,270 All of the TBFVs, with the exception of Powassan virus (POWV), are found in the Old World.271 POWV is found in far eastern Russia and Canada. Deer tick virus (DTV) is a subtype of POWV isolated in New England.813 That significant speciation of POWV in the New World has not yet occurred suggests a relatively recent introduction. The most divergent MBFVs are found in the Old World.271 The earliest lineages of the Aedes clade viruses are thought to originate in Africa; only DENV and YFV are now found in the New World.271 Finally, the distribution of the NKV viruses also appears consistent with an Old World origin. Viruses that infect bats are found in either the Old or New World, whereas those associated with rodents occupy restricted niches in the New World. It is possible that bats played an important role in the introduction of these viruses into the New World.248
Inspection of the phylogenic trees for MBFV and TBFVs revealed striking differences that may reflect distinct biology of the vectors that transmit them. The portion of the tree that includes the TBEV group is highly asymmetric, with a step-wise branching pattern associated with the evolutionary cline of these
viruses.910,911 In contrast, the phylogenic tree for MBFVs is more balanced and does not result in a greater number of branches than predicted by chance. MBFVs evolution appears to involve slow phases punctuated by periods of rapid change and diversification. The last two centuries have been characterized by extensive cladogenesis (change that results in new branches on the phylogenetic tree) for the DENV and JEV complexes.
viruses.910,911 In contrast, the phylogenic tree for MBFVs is more balanced and does not result in a greater number of branches than predicted by chance. MBFVs evolution appears to involve slow phases punctuated by periods of rapid change and diversification. The last two centuries have been characterized by extensive cladogenesis (change that results in new branches on the phylogenetic tree) for the DENV and JEV complexes.
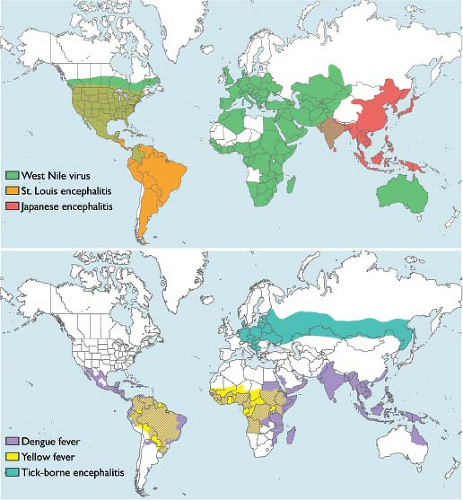 Figure 26.2. Global distribution of flaviviruses. The global distribution of flaviviruses with significant impact on global health. |
Overall, TBFVs appear to have evolved more slowly than the MBFVs (0.56 times the rate of mosquito-borne viruses).911 Several aspects of tick biology may limit the number of replication cycles and dispersal of TBFVs in nature that contribute to clinal pattern and modest rate of evolution: (a) ticks live for relatively long periods (2 to 7 years), (b) ticks feed only three times during their lifespan, (c) ticks may transmit viruses to other ticks during co-feeding, minimizing the importance of the vertebrate host for increasing replication cycles, and (d) ticks are relatively immobile unless carried by a vertebrate host. By comparison, MBFVs are transmitted by vectors with the capacity for wider distributions and are quickly replicated through many cycles in the mosquito vector and vertebrate hosts. MBFVs are found in overlapping distributions (e.g., the four serotypes of DENV), whereas TBFVs characteristically occupy defined and nonoverlapping niches.270
Global Distribution
Flaviviruses are found on six different continents where they are responsible for endemic and epidemic disease each year (Fig. 26.2). The geographic distribution of flaviviruses has proven quite dynamic, enabling emergence in new geographic areas and increased disease incidence.514 For example, since its introduction into the Western Hemisphere in 1999, it took only 4 years for the WNV to spread across the United States, where it is now an endemic pathogen. The contribution of human activity toward the spread of flaviviruses is significant.271 Prior to the development of rapid intercontinental transportation, the movement of flaviviruses between the Old World and New World was uncommon. YFV (and potentially the Aedes aegypti mosquito) were introduced into the Americas during the slave trade 300–400 years ago. Importation of YFV by travelers into nonendemic areas, and DENV and WNV into the New World, has been described.110
Flavivirus Composition and Antigenic Structure
Structure
Flaviviruses are small spherical particles composed of three structural proteins, an ∼11 kb positive-sense genomic RNA, and a lipid envelope. The envelope (E) protein is a ∼53 kD structural protein that functions in multiple steps of the virus life cycle including assembly, budding, attachment to target cells, and viral membrane fusion (reviewed by591). The E protein is also the major target of neutralizing antibodies (reviewed by647,688). The structure of the ectodomain of the E protein has been determined at the atomic level for several flaviviruses.557,559,591,623,647,679,918 Flavivirus E protein is an elongated, type II viral fusion protein composed of three distinct domains connected by short flexible hinges (Fig. 26.3AB). Domain I (E-DI) is an eight-stranded β-barrel located in the center of the E protein molecule. This central domain contains two of the six disulfide bonds present in the E protein structure, as well as a site for the addition of an asparagine-linked (N-linked) carbohydrate. Domain II (E-DII) is an elongated structure that mediates dimerization of E proteins on the mature virion. A highly conserved glycine-rich loop composed of 13 amino acids located at the tip of E-DII is thought to insert into the membranes of target cells.10,98,558 In the context of the dimer, the E-DII fusion loop (E-DII-FL) sits in a hydrophobic pocket formed at the interface of E-DI and domain III (E-DIII). The introduction of mutations into the fusion loop blocks fusion between virions and the membranes of synthetic liposomes.162 For some flaviviruses, E-DII contains a second N-linked glycosylation site. E-DIII adopts an immunoglobulin-like fold at the carboxy-terminus (C-terminus) of the E protein ectodomain and is stabilized by a single disulfide bridge. E-DIII is the portion of the E protein that projects farthest from the surface of the mature virion and is speculated to contain binding sites for cellular factors involved in virus attachment and entry.71,153,455,520,679 Many of the most potent neutralizing antibodies characterized to date recognize epitopes on E-DIII (discussed below). The E protein is tethered to the viral membrane by a helical stem (the stem anchor) and two transmembrane domains.11,591,915
The precursor to membrane protein (prM) is a ∼20 kD protein that facilitates E protein folding and trafficking.501 In addition, interactions with the E protein prevent the adventitious fusion of the virus during egress.321 Virion maturation is regulated by the proteolytic cleavage of prM, which results in the formation of a “pr” protein that is ultimately released from the virion and an ∼8 kD membrane-associated M peptide. The structure of the “pr” peptide has been determined at the atomic level and is composed of seven β strands held together by three disulfide bonds (Fig. 26.4A).466 prM interacts with the E protein near at the tip of E-DII adjacent to the fusion loop.916,917 prM is anchored into the viral membrane via two transmembrane domains.591,915 Recent studies suggest that antibodies specific for prM are commonly produced in vivo.61,191
Flaviviruses assemble on virus-induced membranes derived from the endoplasmic reticulum (ER).353,502,513,861 Virus particles bud into the lumen of these membrane structures as immature virions on which E and prM proteins form heterotrimeric spikes that project from the surface of the virion. Within each spike, the prM protein is located at the tip of the trimer. Immature virions incorporate 60 trimers arranged with icosahedral symmetry (Fig. 26.4B).907,916,917 Transit of the immature virion through the mildly acidic compartments of the trans-Golgi network (TGN) triggers an extensive rearrangement of E proteins on the immature virion; the lower pH induces a structural transition such that E proteins lie flat as antiparallel dimers on the surface of the virion, analogous to the structure of the mature virion discussed below.907 Under acidic conditions, prM remains associated with the fusion loop on this structure and protrudes from the surface of an otherwise smooth virus particle. This pH-dependent conformational change increases the susceptibility of prM for a furin-like serine protease.777 Cleavage of prM is the hallmark of the virion maturation process, and is a required step in the virus life cycle.223 Release of the virion into the neutral conditions of the extracellular milieu results in the dissociation of the pr peptide.466,907 Mature virions are relatively smooth virus particles that incorporate 180 copies of the E protein arranged with an unusual herringbone pseudo-T = 3 icosahedral symmetry425,590,591 (Fig. 26.5A).
Each virion is composed of 30 sets of three antiparallel dimers. In this configuration, E proteins exist in one of three chemically distinct dimer environments defined by their proximity to the two-, three-, or fivefold symmetry axis (Fig. 26.5B).
Each virion is composed of 30 sets of three antiparallel dimers. In this configuration, E proteins exist in one of three chemically distinct dimer environments defined by their proximity to the two-, three-, or fivefold symmetry axis (Fig. 26.5B).
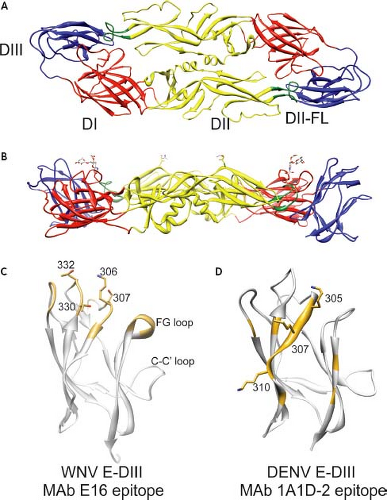 Figure 26.3. Structure of the flavivirus E protein. The envelope (E) proteins of flaviviruses are elongated class II viral fusion proteins composed of three structurally distinct domains. A: Ribbon diagram of the Dengue virus (DENV) E protein dimer as seen from the top; individual domains of each E protein monomer are indicated (domain I, E-DI, red; domain II, E-DII, yellow; and domain III, E-DIII, blue). The fusion loop at the tip of E-DII is shown in green. B: DENV E protein as viewed from the side. The stem anchor connecting the E protein to the viral membrane is not shown. The two N-linked carbohydrate modifications at positions Asn67 and Asn154 are shown. C: Structure of the West Nile virus (WNV) E-DIII highlighting amino acids that form the epitope recognized by the type-specific neutralizing mAb E16. Residues identified in structural studies as antibody contacts are shown in brown; the side chains of residues demonstrated to be critical for antibody binding are shown and labeled. D: Structure of the DENV E-DIII highlighting the epitope recognized by the group-reactive mAb 1A1D-2. Residues identified in structural studies as antibody contacts are shown in brown; the side chains of residues demonstrated to be critical for antibody binding are shown and labeled. We thank Mr. Phong Lee (National Institute of Allergy and Infectious Diseases [NIAID], National Institutes of Health [NIH]) for preparation of the figure. |
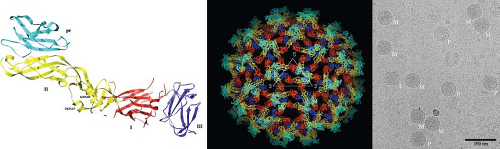 Figure 26.4. The immature flavivirus virion. The structure of prM and the immature flavivirus virion. A: Ribbon representation of the Dengue virus (DENV) pr peptide complexed with the DENV E protein. The pr peptide is shown in cyan. Domains I, II, and III of the E protein are shown in red, yellow, and blue, respectively. B: Surface-shaded representation of the immature DENV virion at neutral pH. C: Cryo-electron microscopy visualization of extracellular DENV reveals the heterogeneity of virions released from mosquito cell cultures. Immature (I) and mature (M) virions are indicated. Partially mature virions (P) characterized by the appearance of smooth and spiky features on the same virion comprise a significant fraction of the virions released from cells. |
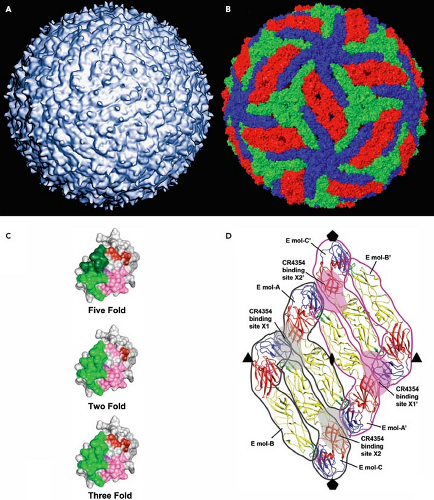 Figure 26.5. The arrangement of E proteins on the mature flavivirus increases the complexity of the antigenic surface of the virus particle. A: Structure of the mature DENV virion. B: Structure of the mature virion highlighting the association of E proteins proximal to the two-, three-, and fivefold symmetry axis of the virion, shown in red, green, and blue, respectively. C: Epitope accessibility varies as a function of the location of a particular E protein on the surface of the pseudo-icosahedral mature virion. Residues important for the binding of mAb E16 are highlighted in green for E proteins of each symmetry environment. The steric conflicts that prevent binding of E16 to E proteins proximal to the fivefold symmetry axis are shown in dark green. Accessibility of amino acids involved in 1A1D-2 binding are shown in pink; steric conflicts that prevent binding to E proteins on the mature virion are shown in red. D: The complex epitope of mAb 4354 composed of multiple E proteins is shown. Individual domains of each E protein monomer are indicated (domain I, E-DI, red; domain II, E-DII, yellow; and domain III, E-DIII, blue). |
The Antigenic Surface
Flaviviruses were first classified according to serologic reactivity.127,188 These early studies generally agreed with approaches that group viruses as a function of genetic relatedness (discussed above, Fig. 26.1).522 Flavivirus-reactive antibodies are classified as a function of their capacity to discriminate between the antigens of viruses within and between related serologic groups of viruses.829 For example, monoclonal antibodies that react with DENV may be type-specific (a single DENV serotype), subcomplex-specific (more than one DENV serotype), complex reactive (all DENV viruses), or flavivirus-group reactive (multiple flaviviruses).322 Type-specific mAbs provided a rapid and specific method for distinguishing between antigenically related viruses.324,572 Furthermore, recent studies indicate that antibodies differentially recognize different genotypes within a given serotype.99,751,852
E Protein Epitopes Recognized by Neutralizing Antibodies
The majority of neutralizing antibodies bind epitopes on the E protein (reviewed in688). Early studies distinguished epitopes on the E protein based on the biochemical and functional properties of mAbs including a capacity to bind and compete for viral antigens, neutralize virus, and inhibit hemagglutination of red blood cells.251,319,322,394,395,411,642,690 An early model of the antigenic structure of the E protein was proposed by Heinz and colleagues that featured three nonoverlapping antigenic domains (A, B, and C); more refined clusters of epitopes within each domain were based on functional differences.286,318 These studies also identified a small number of antibodies that bound the E protein outside of these domains. This advance not only provided a framework to classify antibodies based on their epitope, but also provided context to consider relationships between epitope location and the functional properties of mAbs.689 Antigenic domain A epitopes were recognized by cross- and group-reactive antibodies, domain B epitopes were typically type specific, and domain C epitopes were recognized by subtype-specific mAbs.286,521 These epitopes were subsequently shown to correspond to E-DII, E-DIII, and E-DI, respectively.679
All three domains of the E protein are recognized by neutralizing antibodies, albeit with widely varying potency. Epitopes of some of the most well-characterized antibodies to date are described below, although it should be anticipated that this list will expand as greater insight to the complexity of the antigenic surface of the virion is obtained:
E-DIII-LR
Many of the most potent neutralizing antibodies characterized to date bind epitopes on E-DIII. Potently neutralizing antibodies that bind an epitope on the domain III lateral ridge (E-DIII-LR) of several flaviviruses have been identified.52,150,170,279,627,647,689,708,751 mAb E16 is a type-specific WNV-reactive mAb that neutralizes at picomolar concentrations in vitro and protects mice from lethal challenge when administered 5 days after infection.581,582,627,648 The structure of E16 bound to E-DIII has been determined and revealed a binding footprint composed of four discontinuous loops centered on amino acids of the BC loop and amino-terminal region (positions 306, 307, 330, and 332)624 (see Fig. 26.3C). Type-specific neutralizing antibodies against DENV-2 have been mapped to an epitope composed of multiple regions of the upper lateral surface of E-DIII, including the FG loop.279,360,380,794,795 The binding of many of these antibodies to DIII is also sensitive to amino acid substitutions on the BC loop, C-C′ loop, the amino-terminal region (residue 301), or the A-strand (e.g., residue 304); the latter structure is also recognized by antibodies that neutralize DENV with subcomplex specificity, as discussed below. For example, the highly characterized mAb 3H5 recognizes an epitope composed of residues on both the FG loops and the A strand.795 Mapping experiments with type-specific DENV-1,751 DENV-3,99,852 JEV,891 and TBEV786 antibodies have identified similar epitopes.
E-DIII A-Strand
Antibodies with a pattern of complex and subcomplex reactivity have been mapped to the A-strand of E-DIII.497,671,795,824 mAb 1A1D-2 potently neutralizes DENV-1, DENV-2, and DENV-3, but fails to bind DENV-4 viruses. The structure of mAb 1A1D-2 bound to E-DIII was solved and revealed that this antibody binds an epitope on the A strand and is sensitive to mutation of DENV residues 305, 307, and 310 (see Fig. 26.3D).497 In addition, these studies identified residues in the G strand that play a role in antibody binding, consistent with mapping studies of other complex- and group-reactive antibodies.280,489,751,824
E-DII-FL
Antibodies that bind the fusion loop of E-DII are highly cross-reactive.168,171,629,787 Although mutation of conserved fusion loop residues reduces antibody binding, other adjacent structures also may contribute to the fine specificity of antibody binding and the functional properties of these antibodies.266,629 The structure of the WNV fusion loop-reactive antibody mAb E53 bound to soluble E proteins has been determined.149 Residues shown to be important antibody contacts include those of the fusion loop (residues 104–107, and 109–110) as well as residues of the BC-loop of E-DII.629 The fusion loop epitope is also poorly accessible on the mature virion, as discussed in detail below.629,787
E-DI
Antibodies that bind E-DI have also been characterized. mAb 5H2 is a type-specific antibody that neutralizes DENV-4. Neutralization escape studies mapped 5H2 binding to an epitope that includes residue 174 of E-DI.445 DI-reactive antibodies that bind WNV (E121; residues 175, 191, 193, and 194), DENV2 (mAb DV2–48, residue 177), and TBEV (IC3 and i2, residues D181 and K171, respectively) also have been characterized.338,521,629,794 The recently described WNV mAb CR4354 that binds a complex epitope that includes the linker between E-DI and E-DII will be described in detail below.384
Antibodies That Bind the prM Protein
Antibodies that react with the prM protein have been described.61,121,191,229,844 Generally, these antibodies are characterized by low neutralizing activity in vitro. Recent studies suggest that anti-prM antibodies are elicited frequently in vivo,
and may contribute to the pathogenesis of DENV infection as discussed below.191,685 Human monoclonal antibodies to WNV prM protein have been isolated and mapped to residues V19, T20, T24, and L33.121
and may contribute to the pathogenesis of DENV infection as discussed below.191,685 Human monoclonal antibodies to WNV prM protein have been isolated and mapped to residues V19, T20, T24, and L33.121
Complexities of Antibody Recognition of the Virion
The complex and dynamic arrangement of the E proteins on the surface of flaviviruses complicates an understanding of how antibodies interact with flaviviruses. E proteins exist on the mature virus particle in different chemical environments (Fig. 26.5B; defined in relation to the two-, three-. and fivefold symmetry axes of the pseudo-icosahedral particle), which impacts antibody recognition. Amino acids involved in antibody recognition may be differentially accessible for antibody binding depending on their location on the virus particle.624,629,787 As mentioned above, the WNV mAb E16 binds a cluster of residues on the upper lateral surface of E-DIII.624 However, this epitope is not uniformly accessible for antibody binding on all E proteins on the mature virus; steric constraints imposed by the tight packing of E-DIII on E proteins at the fivefold axis of symmetry prevent antibody binding to these molecules (Fig. 26.5C).383,624 Therefore, although the mature virion incorporates 180 copies of the E protein, a maximum of 120 antibodies physically can bind the virus particle. This is not an unusual feature of this antibody as none of the antibodies studied to date using structural methods appear capable of binding all E proteins on the intact mature virus particle. In fact, the molecular basis for recognition by many antibodies cannot be explained using static models of virion structure.
Epitopes can be Composed of More than One Protein on the Surface of the Virion
Virions contain 180 individual E proteins. To date, most of the well-characterized antibodies are capable of binding monomeric E proteins, indicating their epitopes are composed of residues contained within a single E protein molecule. However, complex epitopes composed of contact residues from adjacent E proteins have been described.168,384,521 mAb CR4354 is a human antibody that binds the hinge between E-DI and E-DII. Mapping studies using recombinant forms of the E protein failed to identify the CR4354 epitope because this antibody was unable to bind soluble forms of the E protein. A loss-of-function substitution at position K136 was defined by neutralization escape studies.850 Cryoelectron microscopic reconstructions of CR4354 Fab fragments bound to the mature virion revealed a complex epitope composed of residues on neighboring E proteins (Fig. 26.5D).384
Incomplete Virion Maturation Impacts Antibody-Mediated Neutralization
Cleavage of prM is a required step in the flavivirus life cycle; mutation of the RRXR/S motif in prM recognized by furin-like proteases renders TBEV noninfectious.223 However, biochemical analysis of preparations of flaviviruses released from cells indicated that a substantial amount of prM may remain uncleaved. Recent studies demonstrate that more than 90% of DENV virions could be precipitated with anti-prM antibodies.368 Electron microscopy studies identified virus particles with structural features of both mature and immature virions (hereafter referred to as “partially mature virions”) (Fig. 26.4C).653 Several lines of evidence suggest that partially mature virions are infectious. Virions produced in the presence of ammonium chloride display a reduced sensitivity to inactivation when exposed to acid, presumably because pH-mediated changes in the conformation of E protein are reversible when complexed with prM.285 In addition, the carbohydrate on prM can mediate attachment of the lineage II 956 strain of WNV (which lacks an N-linked carbohydrate on the E protein) onto cells expressing the c-type lectin CD209L.183 Although these studies demonstrate that virions containing prM may be infectious, the stoichiometric requirements of prM cleavage have not yet been determined.
The presence of partially mature secreted virions impacts antibody recognition in at least two ways. Increasing the efficiency of virion maturation resulted in a marked reduction in the sensitivity of WNV to neutralization by antibodies that bind several structurally distinct epitopes, including the DI-LR and DII-FL epitopes recognized by mAbs E121 and E53, respectively.607 Conversely, decreasing the extent of virion maturation enhanced neutralization by these mAbs. An analysis of the sensitivity of polyclonal antibody elicited by vaccination with two distinct candidate WNV vaccines revealed maturation state-dependent changes in neutralization potency in roughly half the volunteers.607 The structural basis for this pattern of recognition has been investigated.149 mAb E53 does not efficiently bind the E protein on mature virions due to poor accessibility of the fusion loop epitope on the mature virus particle. In addition to modulating the potency of neutralizing antibodies, uncleaved prM on infectious virus particles may interact directly with antibodies, resulting in enhanced infection of Fcγ-receptor bearing cells in vitro and in vivo as discussed below.61,191,347,685,912
Impact of Structural Dynamics of the Virus Particle
Flaviviruses present a complex and dynamic antigenic surface to the immune system that is not fully captured by the static models of virion structure. It has long been appreciated that proteins are in constant motion and sample an ensemble of conformations at equilibrium.84 Proteins incorporated into virus particles also are structurally dynamic.361,888 Virus “breathing” has been demonstrated for several unrelated classes of viruses,88,459 and may affect antibody recognition.467,905 As an example, the accessibility of the A-strand epitope on the mature virion is limited by steric constraints arising from the arrangement of E proteins on the virus particle. The binding of Fab fragments of the subcomplex-reactive A-strand–specific mAb 1A1D-2 to mature DENV virions was shown to be temperature dependent; significant Fab binding was observed only after incubation at 37°C.497 Cryoelectron microscopic reconstruction of mature DENV bound by the 1A1D-2 FAb revealed significant changes in the arrangement and orientation of E proteins on the surface of the virus particle. These results suggested that the binding of 1A1D-2 stabilized the E proteins in a state distinct from the herringbone arrangement found on mature virions. More recent studies suggest the impact of viral dynamics on antibody-mediated neutralization is widespread among antibodies of differing specificity. Analysis of the neutralizing activity of a panel of mAbs specific for structurally distinct epitopes revealed a time- and temperature-dependent aspect of neutralization of WNV and DENV attributed to changes in epitope accessibility arising from the dynamic
motion of E proteins on the virion.210 Given sufficient time, even epitopes that are poorly accessible in all three symmetry axes of the mature virion may support some level of neutralization. The scope of the structural conformations sampled by flaviviruses at equilibrium is not yet understood. Changes in the configuration of E proteins on dynamic virions have the potential to affect several aspects of antibody binding (functional affinity, bivalency, antibody binding orientation), and thus, complicates our understanding of the antigenic surface of the flavivirus virion.
motion of E proteins on the virion.210 Given sufficient time, even epitopes that are poorly accessible in all three symmetry axes of the mature virion may support some level of neutralization. The scope of the structural conformations sampled by flaviviruses at equilibrium is not yet understood. Changes in the configuration of E proteins on dynamic virions have the potential to affect several aspects of antibody binding (functional affinity, bivalency, antibody binding orientation), and thus, complicates our understanding of the antigenic surface of the flavivirus virion.
Antibodies to NS1 Can Protect In vivo
NS1, a protein that is absent from the virion, is secreted at high levels into the extracellular environment during flavivirus infection, predominantly as a hexamer,238 with significant accumulation (up to 50 μg/ml) in the sera of DENV-infected patients.6,28,470,906 In addition, soluble NS1 can bind back to the plasma membrane of cells through an interaction with specific sulfated glycosaminoglycans.29 Furthermore, NS1 is expressed directly on the surface of infected cells, possibly via glycosyl phosphatidyl inositol (GPI) linkage,354 lipid raft association,621 or through an as-yet undefined mechanism. Several groups also have generated nonneutralizing, yet protective mAbs against NS1.155,156,194,230,323,665,720,721,723,724,725 Therefore, protection against flavivirus infections in vivo does not always correlate with neutralizing activity in vitro.93,690,722 Beyond direct virus neutralization, antibody binding to virions or virus-infected cells can trigger protective Fc-dependent antiviral activities through complement activation or Fc-γ receptor–mediated immune complex clearance mechanisms. Fc-γ receptors can activate or inhibit immune responses depending on their cytoplasmic domain and association with specific signaling molecules.617 A requirement for Fc effector function has been established for protective anti-NS1 mAbs. NS1 is expressed on the cell surface or secreted into the extracellular space and antagonizes complement control of flavivirus infection by binding the negative regulator factor H or by promoting C4 degradation.24,154 Passive transfer of mAbs against NS1 can protect mice against lethal infection by WNV and YFV,155,722 and this requires an intact Fc moiety.725 Mechanistic studies using immunodeficient mice demonstrate that protective anti-NS1 mAbs recognize cell surface–associated NS1 and trigger Fc-γ receptor–dependent phagocytosis and clearance of WNV-infected cells.156
Clinical and Pathologic Syndromes of the Flaviviruses
Dengue Virus
History, Global Distribution, and Epidemic Cycle
The natural cycle of epidemic Dengue virus (DENV) infection is between the mosquito vector (Aedes albopictus or Aedes aegypti) and humans (Fig. 26.6). After mosquito inoculation, DENV infection causes a spectrum of clinical disease ranging from self-limited Dengue fever (DF) to a life-threatening hemorrhagic and capillary leak syndrome (Dengue hemorrhagic fever [DHF]/Dengue shock syndrome [DSS]). Globally, there is significant diversity among DENV strains, including four distinct serotypes (DENV-1, DENV-2, DENV-3, and DENV-4) that differ at the amino acid level in the viral envelope proteins by 25% to 40%. DENV causes an estimated 25 to 100 million infections and 250,000 cases of DHF/DSS per year worldwide, with 2.5 billion people at risk.298,562
Although a dengue-like syndrome may have occurred in China several times during the first millennium AD, the initial description of a DENV epidemic is attributed to Benjamin Rush, a physician in Philadelphia, in his article reporting a febrile outbreak in 1780.694 Primary DENV infection and epidemics were common in North America, the Caribbean, Asia, and Australia during the 18th and 19th centuries, presumably due to the widespread ecology of the mosquito vectors. During World War II, DENV spread to and through Southeast Asia. Troop movement and the destruction of the environment and human settlements are believed to have promoted the spread of DENV and their mosquito vectors throughout Southeast Asia and the Western Pacific.433 Since 1950, the number of people infected has risen steadily, such that today DENV is the leading arthropod-borne viral disease in the world. With the spread and co-circulation of multiple DENV serotypes, secondary infection with heterologous serotypes and epidemic DHF/DSS emerged 50 years ago in Southeast Asia,310 and more recently in the Americas in 1981420 and South Asia in 1989.551 Since the 1950s, epidemics involving thousands of people with multiple DENV serotypes and strains occur annually in multiple parts of the world, including the Americas, Asia, Africa, and Australia, in essence wherever the primary mosquito vector Aedes aegypti is present. Indeed, after a recent outbreak of DENV
in Key West Florida in 2009, a serosurvey conducted by the Centers for Disease Control and Prevention (CDC) reported that 5.4% of households had evidence of recent DENV infection.1 As a reflection of this, the global incidence of DHF/DSS has increased more than 500-fold, with more than 100 countries affected by outbreaks of dengue.442
in Key West Florida in 2009, a serosurvey conducted by the Centers for Disease Control and Prevention (CDC) reported that 5.4% of households had evidence of recent DENV infection.1 As a reflection of this, the global incidence of DHF/DSS has increased more than 500-fold, with more than 100 countries affected by outbreaks of dengue.442
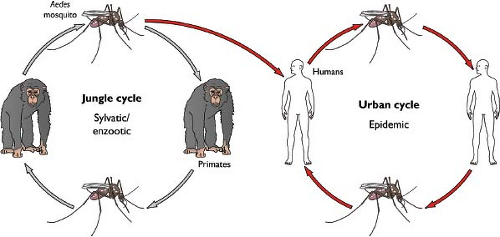 Figure 26.6. Life cycle of Dengue virus (DENV). DENV circulates in nature in two relatively distinct transmission cycles vectored by Aedes sp. mosquitoes. DENV infection of humans results in a sufficiently high viremia to support infection of feeding mosquitoes; transmission cycles of DENV do not require an enzootic amplifying host. DENV may also replicate in a sylvatic cycle. Although incompletely understood, the contribution of sylvatic strains of DENV to human infections appears minimal. |
DENV Diversity
Globally, there is significant diversity among DENV strains. The four serotypes of DENV (DENV-1, DENV-2, DENV-3, and DENV-4) are genetically distinct but cause similar diseases and share epidemiologic features. All DENV strains are members of the Dengue antigenic complex; inclusion of a strain as DENV is based on antigen cross-reactivity, sequence homology, and genome organization.120 The four serotypes of DENV were historically distinguished by limited cross-neutralization or hemagglutination inhibition using serum from infected individuals. Subsequent sequencing analysis revealed that individual serotypes of DENV can differ from one another at the amino acid level significantly, with 30% to 40% variation in the viral envelope proteins. However, within a given serotype, amino acid homology is much greater, with conservation levels at approximately 90% or higher. Therefore, individual DENV serotypes (e.g., DENV-1 versus DENV-4) vary far more than distinct viruses in Japanese encephalitis serocomplex (e.g., WNVs and JEVs vary by 10% to 15% at the amino acid level), which has led some to consider DENV as a group of four different viruses that are linked by serology, epidemiology, and disease pathogenesis. Differences in severity associated with individual serotypes or particular sequences of serotypes in sequential infection have been observed, and it still is unclear whether some serotypes are inherently more pathogenic than others. DENV-2 viruses have been commonly associated with DHF/DSS,36,112,821 as are DENV-1 and DENV-3 viruses.273,314,551 In comparison, DENV-4 appears more commonly to be clinically mild, although it can cause severe disease.619
Genetic variation of DENV, however, is not limited to serotype. Geographic variants within a serotype were initially identified by RNase fingerprint assays.678,847 Subsequently, nucleic acid sequencing confirmed differences within each serotype, allowing for classification of genotypes that vary further by up to approximately 6% and 3% at the nucleotide and amino acid levels, respectively.337,681 DENV genotype classification was originally defined by sequence variation within a given genomic region (e.g., E and NS1 genes). More recent analysis has used high-throughput full genome sequencing technologies to assign phylogenetic classification. Although there remains some dissonance among investigators, most classification schemes include five DENV-1 genotypes, four or five DENV-2 genotypes, four DENV-3 genotypes, and two or three DENV-4 genotypes.682,874 Beyond serotype and genotype, two further types of DENV complexity should be mentioned: strain variation and quasispecies. DENV strain variation refers to the limited change that occurs among individual isolates; this was classically described as within a serotype, although as DENV continues to emerge and evolve, variation now occurs within a genotype. Strain variation within a genotype may be functionally important, as it can affect antibody neutralization, presumably due to changes at key sites within exposed epitopes.99,794,852,925
In addition to serotype, genotype, and strain variation, DENV has the capacity to accumulate variation rapidly within an individual host. Viral quasispecies comprises a cloud of variants that are genetically linked through mutation. It is observed during infection by many RNA viruses (e.g., hepatitis C virus [HCV], human immunodeficiency virus [HIV], and influenza) and creates diversity that allows a viral population to adapt rapidly to dynamic environments and evolve resistance to immune responses, vaccines, and antiviral drugs.450,848 DENV exists as a collection of highly similar variants forming a quasispecies869 by virtue of its error-prone NS5 polymerase, which has an estimated mutation rate of 103 to 105 substitutions per nucleotide copied per round of replication.137,836 Preliminary studies suggest that genetic diversity is greater in the structural proteins, which may have less constraint to maintain integral functions. The study of genetic and intrahost diversity for DENV is still in its relative infancy, and thus more analysis is warranted to define how mutation and variation impact fitness, tropism, and resistance.
Clinical Features of Acute DF: Primary DENV Infection
DENV infection of humans after mosquito inoculation causes a spectrum of clinical disease ranging from inapparent disease (∼50% of infections37,112,224), self-limited dengue fever (DF) to severe DHF and DSS. A classical presentation of DF is an abrupt onset of a debilitating febrile illness characterized by headache, retroorbital pain, myalgias, arthralgias, and a maculopapular rash that occurs after a 2- to 7-day incubation period after mosquito inoculation.700 Some individuals experience severe bone and joint pain (“break-bone fever”) and develop petechial hemorrhages that are associated with mild to severe thrombocytopenia. There is no specific constellation of signs or symptoms to differentiate DF from other acute flu-like viral syndromes, so a health care provider must have a high index of suspicion for diagnosis in the setting of the appropriate epidemiology. DF also may present in a less classical form as an undifferentiated febrile illness with rash along with mild upper respiratory symptoms (cough, pharyngitis, rhinitis), particularly in children. DF is usually self-limited, lasting 1 to 2 weeks, although some (up to 25% of hospitalized patients) experience a prolonged postinfectious fatigue and depression syndrome that can persist for weeks, akin to that seen after Epstein-Barr virus (EBV) infection and mononucleosis.739 Because of the debilitating fever and musculoskeletal symptoms, the morbidity toll is high in clinically apparent DF, whereas the mortality rate is exceedingly low. Primary DF usually occurs during the initial DENV infection of an individual, with the exception of infants from immune mothers that have acquired antibodies transplacentally.
Clinical Features of DHF/DSS: Secondary and Infant DENV Infection
The incidence of the most severe form of DENV disease, DHF/DSS, varies considerably between primary and secondary infections. A secondary DENV infection results when a person previously infected with one serotype is exposed to a different serotype, and is the single most important risk factor for severe dengue disease.112,224,294,305 Epidemiologic data in Thailand has shown greater than 10-fold higher rates of DHF/DSS during secondary compared to primary infection of children.843 It should be pointed out that even during secondary infection, DHF/DSS is quite rare, with only 0.5% of
secondary infections progressing to the most severe forms of dengue disease. DHF/DSS is characterized by rapid onset of capillary leakage accompanied by thrombocytopenia and mild to moderate liver damage, reflected by increases in serum levels of hepatic enzyme (e.g., aspartate aminotransferase [AST] and alanine aminotransferase [ALT]).301 DHF/DSS usually occurs as a second phase of the illness, after a short period of defervescence from the initial fever. Hemorrhagic manifestations are observed in a subset of DHF/DSS cases and include petechiae, epistaxis, gastrointestinal bleeding (hematemesis or melena), menorrhagia, and a positive tourniquet test. Use of the term hemorrhagic fever instead of dengue capillary-leak syndrome has led many to anticipate that bleeding is the greatest threat. Rather, fluid loss into tissue spaces with hemoconcentration and hypotension can result in shock, which carries the highest risk of mortality.612 From a diagnostic standpoint, an elevated hematocrit and upper abdominal ultrasonogram showing a thickened gall bladder wall, hepatomegaly, ascites, or pleural effusions are evidence of fluid shifts associated with a capillary leak syndrome.
secondary infections progressing to the most severe forms of dengue disease. DHF/DSS is characterized by rapid onset of capillary leakage accompanied by thrombocytopenia and mild to moderate liver damage, reflected by increases in serum levels of hepatic enzyme (e.g., aspartate aminotransferase [AST] and alanine aminotransferase [ALT]).301 DHF/DSS usually occurs as a second phase of the illness, after a short period of defervescence from the initial fever. Hemorrhagic manifestations are observed in a subset of DHF/DSS cases and include petechiae, epistaxis, gastrointestinal bleeding (hematemesis or melena), menorrhagia, and a positive tourniquet test. Use of the term hemorrhagic fever instead of dengue capillary-leak syndrome has led many to anticipate that bleeding is the greatest threat. Rather, fluid loss into tissue spaces with hemoconcentration and hypotension can result in shock, which carries the highest risk of mortality.612 From a diagnostic standpoint, an elevated hematocrit and upper abdominal ultrasonogram showing a thickened gall bladder wall, hepatomegaly, ascites, or pleural effusions are evidence of fluid shifts associated with a capillary leak syndrome.
Whereas DHF/DSS occur largely after secondary infection by a different DENV serotyope in children and adults,273 in infants younger than age one born to dengue-immune mothers, primary infection can cause severe DHF/DSS.305,356,764 In clinical studies, maternal anti-DENV neutralization antibody titers and age of the infant correlated with disease. The actual age at which DHF/DSS occur in infants (peak at 7 months) corresponds to the age at which maximum enhancing activity for DENV infection in primary monocytes is observed in vitro.405 Severe clinical manifestations of DHF/DSS are more prevalent in infants311 and there is an approximately fourfold higher mortality rate compared to other age groups.373 Infants represent approximately 5% of children hospitalized with DHF/DSS in many parts of Southeast Asia.140,273,303 The more prevalent or severe clinical manifestations associated with infant DHF/DSS include seizures, hepatic dysfunction, thrombocytopenia, high-grade fever, diffuse rash, peripheral edema, ascites, and frank shock.356
Although it is not fully accepted by the field, some clinical studies have suggested that severe DENV infection also can have neurologic manifestations including transverse myelitis, Guillain-Barre syndrome, encephalitis, and encephalopathy,769,841 occurring in as many as 1% to 6% of DHF/DSS cases.122,325 In contrast to other encephalitic flaviviruses (e.g., JEV, WNV, or tick-borne encephalitis viruses), DENV historically has not been considered as neurotropic. However, the discovery of DENV and anti-DENV immunoglobulin M (IgM) in the cerebrospinal fluid of patients with encephalopathy suggests that it may be capable of causing central nervous system (CNS) infection as part of a severe DHF/DSS syndrome, at least in a subset of individuals.507,769 In support of this, focal imaging abnormalities have been detected in brain MRI scans of DENV-infected patients.122,872 Although these results are suggestive, bona fide DENV encephalitis and CNS disease may not be fully accepted until its antigens are reliably detected in the brains of encephalopathic patients and a more complete understanding of the molecular determinants for neurotropism is acquired.
Pathologic Features of DHF/DSS
Although DENV is the leading mosquito-borne transmitted viral infection in the world, there are few detailed autopsy series of patients who succumbed to DHF/DSS, and fewer performed with newer molecular techniques and markers. Detailed histopathologic studies that might inform a basic understanding of DENV pathogenesis are rare because much of the lethal disease occurs in regions lacking sophisticated laboratory infrastructure, highly trained personnel, and repositories for long-term tissue storage. Forensic studies also are complicated by lack of standardization of histologic procedures and variation in the quality of specimen preparation and storage.
A recent summary of the autopsy literature from a total of 160 fatal DHF/DSS cases occurring primarily in children and adolescents was published.531 Pathologic findings in the liver of DHF/DSS cases include centrilobular necrosis, changes in fatty tissue, inflammatory leukocyte infiltration, and Kupffer cell hyperplasia.70,113 Gross macroscopic examination revealed multiple hemorrhagic foci. Microscopic analysis has shown increased inflammatory infiltrates around the portal vessels, sinusoidal congestion, small hemorrhages, midzonal hepatocyte necrosis, and microvesicular steatosis.228,293 In other tissues (spleen or lung) hemorrhage, tissue edema, and plasma leakage have been observed.50
A key to understanding the pathogenesis of severe DENV infection is defining cellular tropism of infection, which could influence the host inflammatory response that results in the capillary leakage syndrome. Autopsy series have shown the presence of DENV antigen or nucleic acid in cells of the skin, liver, spleen, lymph nodes, kidney, lung, thymus, or brain.38,50,165,293,377,392,553,692 However, several of these studies used in situ hybridization or reverse transcriptase polymerase chain reaction (RT-PCR) based assays, and thus have not definitively shown that infectious virus is produced in a given cell of a target tissue. In severe DENV cases, infectious virus can be reliably isolated from blood, lymphoid tissues, and the liver, although the cellular source of the virus remains controversial. Studies in humans, nonhuman primates, and small animal models support a role for infection of myeloid cells (blood monocytes, tissue macrophages, Kupffer cells), and possibly other cells including hepatocytes296 and endothelial cells.50,912
Yellow Fever Virus
History, Global Distribution, and Epidemic Cycle
Several recent excellent reviews have described the epidemiology and historical details of yellow fever virus (YFV) infection.41,222,246 YFV, the causative agent of yellow fever virus (YFV), was first isolated (strain Asibi) in 1927 after inoculation of a rhesus monkey with the blood of a patient from Ghana.789 YFV originated in Africa, was imported into the Americas during the slave trade, and had the first reported epidemic in the Yucatan in 1648.45 Historically, large epidemics of YF disease occurred beyond these regions and were described in the 17th through 20th centuries as far north in the Americas as Canada, as well as in parts of Europe including Spain, Italy, France, and England.566 Despite the presence of an effective vaccine (17D strain) that was developed in 1937 by Max Theiler and colleagues,820 with more than 500 million doses administered to humans,246 YFV infection has remained a public health threat in restricted parts of the world. Currently, YFV is endemic in the tropical regions of Africa and the Americas, infects humans and
nonhuman primates, and is transmitted by mosquitoes including Aedes aegypti. The World Health Organization (WHO) estimates an incidence of 200,000 cases per year, leading to about 30,000 deaths, with the majority occurring in sub-Saharan Africa.890 Overall, 44 countries in Africa and the Americas are considered within the modern YFV endemic zone, with almost 900 million people at risk of infection.222,246
nonhuman primates, and is transmitted by mosquitoes including Aedes aegypti. The World Health Organization (WHO) estimates an incidence of 200,000 cases per year, leading to about 30,000 deaths, with the majority occurring in sub-Saharan Africa.890 Overall, 44 countries in Africa and the Americas are considered within the modern YFV endemic zone, with almost 900 million people at risk of infection.222,246
The sylvatic or jungle cycle of YFV in which transmission occurs between mosquitoes and wild monkeys explains why extensive vaccination campaigns have reduced but not eradicated infection. In East Africa, YFV infection is maintained enzootically in monkey transmission cycles in the jungle with the Aedes africanus mosquito vector. Periodically, infection may cross into humans during an intermediate savannah cycle, with transmission by several different Aedes mosquito species (e.g., Aedes bromeliae). Indeed, in the Americas, most cases appear to be a result of humans intruding on the jungle cycle of YFV.47 Epidemic YFV infection (human–mosquito–human cycle) ensues in urban or domestic areas with Aedes aegypti as the principal mosquito vector. Rapid urbanization in Africa and the Americas with population shifts from rural to urban settings combined with the collapse of mosquito eradication programs has allowed the A. aegypti vector to repopulate many parts of the world, and caused YFV to be classified as a reemerging threat for humans.246
YFV Diversity
YFV does not belong to an antigenic subgroup based on plaque reduction neutralization assays,120 but shows greater genetic relationship to other African flaviviruses including Banzi, Zika, Wesselsbron, and Bouboui viruses. Indeed, cross-protection against YFV infection in monkeys has been shown after immunization with some of these related viruses.326 Although there is only one serotype of YFV, there is significant diversity within the genus. Seven genotypes have been proposed including two West African genotypes, a single Central/South African genotype, two East African genotypes, and two South American genotypes.222 These genotypes of YFV were originally defined based on nucleotide variation of greater than 9% in the prM, E, and 3′ UTR gene regions601,858 and have been confirmed with full-genome sequencing of YFV isolates.851 Phylogenic studies suggest that YFV originated in East or Central Africa and was introduced subsequently into West Africa and South America.600,601 Beyond genotypes, sequence analysis of 79 YFV strains isolated from 1935 to 2001 in Brazil revealed further strain divergence into clades that differ at the nucleotide and amino acid level by up to 7% and 5%, respectively.842 The physiologic basis for genotype-specific amino acid variation between YFV isolates remains uncertain, although it is likely that selection confers a phenotypic advantage in a given host.
Clinical Features of YFV Infection
In humans, YFV infection causes a variable clinical syndrome ranging from no symptoms, to mild febrile flu-like illness, to fulminate and possibly fatal disease. Approximately 15% of people who become infected develop severe visceral disease, and in this group there is a 20% to 50% case fatality rate.564 Symptoms occur within 3 to 6 days of mosquito inoculation and include an abrupt onset of fever, chills, myalgia, back pain, and headache or the first “period of infection,” which usually lasts 3 days and corresponds to peak viremia. During this phase, individuals are infectious to mosquitoes. In some, this stage may be followed by a short “period of remission,” with defervescence and improvement of clinical signs and symptoms. Shortly after, in a subset (20%) of patients, fever and symptoms worsen (“period of intoxication”) with vomiting, epigastric pain, and jaundice (which gives yellow fever its name); this is associated with YFV replication in the liver, an absence of viremia, and measurable anti-YFV antibodies in serum. As time progresses, severe YFV infection evolves into a hemorrhagic fever characterized by severe hepatitis, renal failure, hemorrhage, shock, and multiorgan failure. A bleeding diathesis manifests with melena, hematemesis, epistaxis, ecchymosis, menorrhagia, petechial hemorrhages, and blood oozing from mucous membranes. Renal failure is associated with an abrupt decrease in urine output and with albuminuria. Laboratory tests show leukopenia, thrombocytopenia, and a coagulopathy. Death occurs on the 7th to 10th day of illness and is preceded by hemodynamic and cardiovascular instability, acute liver failure, hypothermia, hypoglycemia, and coma. For those individuals surviving severe YFV infection, convalescence is prolonged with hepatitis and associated constitutional symptoms persisting sometimes for months.
Pathologic Features of YFV Infection
Macroscopic gross pathology of tissues from YFV infection autopsy studies show an enlarged and icteric liver and edematous and enlarged kidneys and heart. Microscopic pathologic analyses of the liver reveal six major features,246,408,567 which occur primarily during the last “period of intoxication”: (a) eosinophilic degeneration of hepatocytes and Kupffer cells; (b) midzonal hepatocellular swelling and necrosis, with sparing of the cells in the portal area; (c) the presence of Councilman bodies coincident with hepatocyte cell death; (d) absence of leukocyte inflammatory infiltrates; (e) microvesicular fatty changes and lipid accumulation, likely secondary to decreased apoprotein synthesis by hepatocytes; and (f) retention of the reticulin structure. YFV antigen and RNA are demonstrable in hepatocytes by immunohistochemistry or in situ hybridization,187 and this coupled with the absence of inflammation, suggests that the cell death is mediated directly by virus infection, likely via apoptotic mechanisms.567,666
In the kidney, severe eosinophilic degeneration and a microvesicular fatty change of renal tubular epithelium are observed, analogous to that seen in the liver. Viral antigen can be detected by immunohistochemistry in renal tubular cells.187 Glomerular damage and albuminuria with changes in the basement membrane and degeneration of cells lining Bowman’s capsule may be due to direct viral injury567 or secondary to decreased blood flow during the sepsis syndrome.246 The spleen shows an overall loss of lymphocytes, hyperplasia of the follicle, appearance of large mononuclear tissue histiocytes, and significant degeneration of cells with accumulation of fragmented nuclei.409 In monkeys, necrosis of B-cell follicular areas of the spleen is more apparent.568 In the heart, myocardial cells also undergo apoptotic changes as in other organs, in the absence of a significant cellular inflammatory response. Patchy lesions have been described in sinoatrial (SA) node and bundle of His,495 which could explain the paradoxical bradycardia (Faget’s sign) and late cardiac death, observed in some severe YFV cases.
Hemorrhagic manifestations and damage to and plasma leakage from capillaries are characteristic findings of severe
YFV infection.567 The bleeding manifestations are attributed to decreased synthesis of vitamin K–dependent coagulation factors by the injured liver, disseminated intravascular coagulation, and reduced platelet numbers and function. Beyond direct bleeding, there is additional vascular dysfunction, with pleural and peritoneal effusions, and edema of several other organs, including the brain. At present, the precise pathogenesis of the vascular leakage syndrome associated with YFV remains unknown, although highly elevated levels of proinflammatory and vasoactive cytokines are observed.814
YFV infection.567 The bleeding manifestations are attributed to decreased synthesis of vitamin K–dependent coagulation factors by the injured liver, disseminated intravascular coagulation, and reduced platelet numbers and function. Beyond direct bleeding, there is additional vascular dysfunction, with pleural and peritoneal effusions, and edema of several other organs, including the brain. At present, the precise pathogenesis of the vascular leakage syndrome associated with YFV remains unknown, although highly elevated levels of proinflammatory and vasoactive cytokines are observed.814
West Nile Virus
History, Global Distribution, and Epidemic Cycle
West Nile virus (WNV) was first isolated in 1937 in the West Nile district of Uganda from a woman with an undiagnosed febrile illness.766 Historically, WNV caused sporadic outbreaks of a mild febrile illness in regions of Africa, the Middle East, Asia, and Australia. Indeed, in the 1950s, detailed studies of WNV showed recurrent outbreaks in Israel66,260 and high levels of seroconversion in adults from Egypt352,549; these outbreaks and others in Africa generally were not associated with severe human disease. However, in the 1990s, the epidemiology of infection changed. New outbreaks in Eastern Europe were associated with higher rates of neurologic disease.348 In 1999, WNV entered North America, and caused seven human fatalities in the New York area as well a large number of avian and equine deaths. Over the last decade, WNV has spread to all 48 of the lower United States as well as to parts of Canada, Mexico, the Caribbean, and South America. Because of the increased range, the number of human cases has continued to rise: in the United States between 1999 and 2012, 36,500 cases were confirmed and associated with 1,500 deaths (http://www.cdc.gov/ncidod/dvbid/westnile/surv&control.htm).
WNV cycles in nature between Culex mosquitoes and birds, but also infects and causes disease in humans, horses, and other vertebrate species. (Fig. 26.7) Ticks also have been implicated as having a minor role in transmission in some parts of the world,510 although few isolates have been obtained. Although its enzootic cycle is overwhelmingly between mosquitoes and birds, with vertebrate species serving as “dead-end” hosts because of low-level and transient viremia, nonviremic transmission of WNV between co-feeding mosquitoes330 suggests that vertebrates could act as reservoirs for mosquito infection. Most (∼85%) human infections in the Northern Hemisphere occur in the late summer, with a peak number of cases in August and September. This reflects the seasonal activity of Culex mosquito vectors and a requirement for virus amplification in the late spring and early summer in avian hosts. In warmer parts of the world, virtually year-round transmission has been observed. Although more than 100 avian species are susceptible to WNV infection, in the United States, some are particularly vulnerable, with a large number of deaths in crows, blue jays, and hawks. The magnitude of dying birds in a community in the early summer often predicts the severity of human or equine disease weeks later.415 Ecologic studies suggest that Culex pipiens, the dominant enzootic (bird-to-bird) and bridge (bird-to-human) vector of WNV in urbanized areas in the northeast and north-central United States, shifts its feeding preferences from birds to humans during the late summer and early fall, coincident with the dispersal of its preferred host, the American robin (Turdus migratorius).393
WNV Diversity
Sequencing and phylogenic analysis of full-length genomes has resulted in a division of WNV strains into four distinct lineages,67,358,447,510 with lineage 1 strains further separated into three clades (1a, 1b, and 1c). This topic has been analyzed in great detail in a recent study.537 Clade 1a comprises isolates from Europe, the Middle East, Russia, and the Americas, and includes all strains from the recent epoch in the United States and Canada. Clade 1b contains the naturally attenuated Australian variant, Kunjin virus, which forms a tight cluster with approximately 2% to 3% difference at the amino acid level from North American WNV strains.719 Clade 1c comprises isolates from India only. Historically, lineage 2 isolates were isolated from sub-Saharan Africa and Madagascar, and generally showed less ability to cause disease in humans and animals54,348; a more recent study suggests that lineage 2 isolates now circulate in parts of Eastern Europe, some of which cause severe disease.226 There are fewer sequenced strains from lineage 3 and 4 WNV, with only one lineage 3 isolate from Austria in 199733 and several lineage 4 isolates510 from Russia between 2002 and 2006. Within a given ecological niche, possibly because of the enzootic cycle, WNV has remarkable genetic stability despite its error-prone RNA-dependent RNA polymerase; full-length sequencing analysis of North American isolates over the past decade
has revealed a rate of approximately five nucleotide and fewer than one amino acid mutation per genome per year, with little geographic subdivision.181,810
has revealed a rate of approximately five nucleotide and fewer than one amino acid mutation per genome per year, with little geographic subdivision.181,810
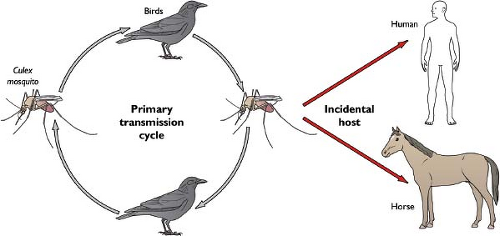 Figure 26.7. Transmission cycle of West Nile virus (WNV). WNV is maintained in nature in an enzootic transmission cycle between mosquitoes and birds. Many vertebrate species, including humans, may also be infected as “dead-end” hosts for WNV. The resulting transient low-level viremia in mammalian hosts does not support sufficient infection of the mosquito vector to continue the transmission cycle. |
Clinical Features of WNV Infection
Seroprevalence studies suggest that most (∼80%) cases are subclinical, without significant symptoms. Among clinical cases, many develop a self-limiting illness that is termed WNV fever. This syndrome begins after a 2- to 14-day incubation period and is characterized by fever accompanied by myalgias, arthralgias, headache, fatigue, gastrointestinal complaints, maculopapular rash, or lymphadenopathy. This nonneuroinvasive form of WNV infection can be severe, as 38% of patients with WNV fever were hospitalized with a mean length of stay of 5.4 days.350 A subset of the symptomatic cases progress to the neuroinvasive forms of WNV infection, including acute flaccid paralysis, meningitis, encephalitis, and ocular manifestations34,742; in many instances, a combination of these syndromes is present. Overall, about 1 in 150 WNV infections result in the most severe and potentially lethal form of the disease. During an epidemic, on a human population scale, the seroconversion rate is approximately 3%588,832 and the attack rate for severe disease during an epidemic is about 7 per 100,000.350 The risk of severe WNV infection is greatest in the elderly.151,603,832 At least two studies have estimated a 20-fold increased risk of neuroinvasive disease and death in those older than 50 years of age.350,603 Persistent movement disorders, cognitive complaints, and functional disability may occur after West Nile neuroinvasive disease. West Nile poliomyelitis-like disease may result in limb weakness and long-term morbidity. Moreover, even patients with apparently mild cases of acute disease have sustained subjective and somatic sequelae following WNV infection. Therefore, the neurologic and functional disability associated with WNV infection represents a considerable source of morbidity in patients long after their recovery from acute illness.740,741,742,743
Although most human WNV infections occur after the bite of an infected Culex mosquito, other routes including transfusion, organ transplantation, and placental and breast milk transmission. In 2002, 23 cases of WNV infection were identified after transfusion of blood products.639 These cases led to the development and implementation of nucleic acid amplification tests, which have been used to identify infected pools or individual blood product samples115,644 and largely prevent transmission by transfusion. Nucleic acid screening of blood donors have not completely eliminated transfusion-transmitted WNV infections, as “breakthrough” infections have occurred, and were attributed to units that had levels of viremia below the sensitivity of the screening assay.116 In addition to transfusion-associated WNV infection, several cases by organ transplantation have been reported.427,428 Because of the relatively low incidence of WNV infection in organ transplantation and risk of false-positives that can occur with wide scale testing, screening is not mandated.
Pathologic Features of WNV Infection
WNV causes encephalitis in several vertebrate species including humans, horses, and birds, by virtue of its ability to infect and cause injury to neurons through direct (viral-induced) and indirect (immune response induced) mechanisms.129 Pathologic observations in humans, however, is limited by the small number of autopsy studies on individuals succumbing to WNV infection. Gross macroscopic examination of organs (brain, lung, kidney, and spleen) tends to be unremarkable.630 Microscopic examination of the brain in humans and other animals reveals histologic changes that are consistent with the clinical disease.221,630 This includes neuronal cell death, activation of resident microglia and infiltrating macrophages, perivascular and parenchymal accumulation of CD4+ and CD8+ lymphocytes and CD138+ plasma cells, and formation of microglial nodules. These lesions, which tend to be patchy in distribution, occur in the brainstem, cerebral cortex, hippocampus, thalamus, and cerebellum.630 In addition, overt meningitis with cellular infiltrates in the meninges can be readily apparent. In some cases, destruction of vascular structures with focal hemorrhage is present, suggestive of a vasculitis; this may be associated with local compromise of the blood–brain barrier.200,868 Immunohistochemical analysis confirms that WNV antigen is present in neurons from multiple regions of the brain, although other cells (e.g., astrocytes or CD11b+ myeloid cells) may be infected to lesser degrees.176,208
In addition, WNV infection can cause a poliomyelitis-like syndrome of acute flaccid paralysis.257,458 Patients show markedly decreased motor responses in the paretic limbs, preserved sensory responses, and widespread asymmetric muscle denervation without evidence of demyelination or myopathy.458 Microscopically, in the spinal cord, an intense inflammatory infiltrate around large and small blood vessels is observed with large numbers of microglia in the ventral horn. Anterior horn motor neurons are targeted by WNV,458,754 and studies suggest that axonal transport from peripheral neurons can mediate WNV entry into the spinal cord and induce acute flaccid paralysis.705
Although most mammalian WNV infections are cleared by the adaptive immune response, persistence in the kidney has been described, albeit infrequently. Hamsters experimentally infected with WNV developed chronic renal infection and viruria for up to 8 months, despite clearance from blood and the appearance of neutralizing antibodies. Although minimal histopathology was reported, WNV antigen staining was detected in the renal epithelium, interstitial cells, and tubules.816 Of interest, these persistent viruses evolved genetically and no longer caused neuroinvasive disease on challenge of naïve animals.895 Analogous to the studies in hamsters, WNV RNA was demonstrated in 5 of 25 urine samples from convalescent humans 1.6 to 6.7 years after the initial infection, although infectious virus was not successfully isolated.599 However, a separate larger study did not detect viral RNA in urine, and thus analysis of additional patient cohorts may be required to better define renal persistence and its significance in humans.254
Persistent WNV infection in the CNS also has been suggested by experimental infection studies in monkeys, hamsters, and mice. In monkeys, WNV persisted at least 5.5 months after initial infection and was isolated in the cerebellum and cerebral subcortical ganglia but had lost its neurovirulence and cytopathic properties.655 In hamsters, persistent WNV RNA and foci of WNV antigen-positive cells were identified in the CNS of hamsters between 28 to 86 days after infection,758 and this was associated with long-term neurologic sequelae. In mice, infectious WNV persisted in the brains of wild-type animals up to 4 months, and viral RNA could be detected at 6 months in up 12% of mice, even in animals with subclinical infection.16 Consistent with this, virus-specific B- and
T-cell immune responses persisted in the CNS of mice up to 4 months after infection.785
T-cell immune responses persisted in the CNS of mice up to 4 months after infection.785
Japanese Encephalitis Virus
History, Global Distribution, and Epidemic Cycle
Japanese encephalitis virus (JEV) is a mosquito-transmitted flavivirus and the prototype virus of the JEV antigenic serocomplex. JEV causes severe neurologic disease, primarily in Asia, where it accounts for about 35,000 to 50,000 cases and 10,000 to 15,000 deaths annually.831 JEV epidemics were originally described in Japan in the 1870s, and the virus was initially recovered in 1935 from the brain of an infected human in Tokyo; this isolate was established as the prototype Nakayama JEV strain.460 Although most human infections are asymptomatic or result in mild symptoms, greater than 50% of the severe clinical cases are fatal or result in devastating long-term neurologic sequelae.736 Moreover, as JEV-induced disease largely occurs in children living in rural areas, it is likely vastly underreported in most regions of Asia.736,768
The enzootic cycle of JEV is between waterbirds (e.g., egrets and herons) and mosquitoes, with pigs also serving as an amplifying host. JEV is transmitted primarily by Culex mosquitoes (principally Culex tritaeniorhynchus) that breed in rice fields and stagnant water. Humans and other vertebrate animals are considered incidental targets and dead-end hosts, as they do not produce a viremia sufficient to infect mosquitoes. Two epidemiologic patterns are observed: in northern temperate areas JEV infections occur during the summer months, whereas as in tropical climates, year-round transmission of JEV has been described.267
Globally, despite the introduction of several inactivated and live-attenuated vaccines (see Vaccine section below), JEV remains the most important cause of arthropod-transmitted viral encephalitis. Disease caused by JEV is widely distributed in Asia, with outbreaks historically occurring in Japan, China, Taiwan, Korea, the Philippines, India, parts of Southeast Asia, and the far-eastern region of Russia. Although cases in China appear to be declining, possibly due to large-scale vaccination campaigns, epidemic activity in India, Nepal, and other parts of Southeast Asia appears to be escalating. More recently, JEV has been described in Pakistan, Papua New Guinea, and Australia, suggesting that its geographic range may be expanding.312,313
JEV Diversity
Phylogenic analysis suggests that JEV evolved from an ancestral flavivirus in Africa within the last few centuries.268 Based on sequence analysis primarily of the viral structural genes, JEV was initially classified into one single serotype with four distinct genotypes (I–IV),145,146,835 with as much as 12% variation at the nucleotide level. These divisions have been confirmed by full-length genome sequencing on a subset of isolates. Genotype I includes isolates from Thailand, Cambodia, Korea, China, Japan, Vietnam, Taiwan, and Australia from 1967 to the present. Genotype II includes strains from Thailand, Malaysia, Indonesia, Papua New Guinea, and Australia from 1951 to 1999. Genotype III includes isolates recovered from mostly temperate areas of Asia including Japan, China, Taiwan, the Philippines, and the Asian subcontinent between 1935 and the present. Finally, genotype IV includes strains from Indonesia that were isolated only in 1980 and 1981. More recently, a fifth, more divergent genotype (V) has been proposed based on full-genome sequencing of a 1952 isolate from a patient in the Muar region Malaysia.560 This strain has approximately 20% and 9% nucleotide and amino acid divergence, respectively, and shows significant variation with respect to neutralization by JEV-specific monoclonal antibodies.315
Because genotypes I and III largely occurred in epidemic regions and genotypes II and IV were associated with endemic transmission, differences in strain virulence were hypothesized to explain the epidemiologic patterns of JEV.146 However, as the geographic range of JEV has expanded, there are now several examples in which strains of individual genotypes cause either epidemic or endemic disease depending on the region or country.772
Clinical Features of JEV Infection
In humans, the JEV infection can be asymptomatic or produce a range of clinical syndromes including a mild nonspecific febrile illness, aseptic meningitis, seizures, encephalitis, and poliomyelitis-like flaccid paralysis. Disease onset usually begins with a 1- to 2-week period of flu-like symptoms including headache, fever, cough, and upper respiratory symptoms, as well as gastrointestinal complaints such as nausea, vomiting, and diarrhea. In infants and young children the disease can progress rapidly as the virus invades the CNS and infects and injures neurons. CNS invasion is heralded by nuchal rigidity, photophobia, and altered mental status. JEV infection in the CNS can share features with Parkinson’s disease including mask-like facies, hypertonia, tremor, and cogwheel rigidity. Other CNS symptoms include seizures (more common in children than adults), ataxia, involuntary movements (e.g., choreoathetosis, facial grimacing, and lip-smacking), and cranial nerve palsies. Associated with this are elevated white blood cell counts and pressure in the cerebrospinal fluid (CSF) and abnormal electroencephalography (EEG) examinations. Imaging studies in the brain have revealed thalamic and basal ganglia abnormalities during the acute phase of disease.768 Upper rather than lower extremity paralysis is more common, and lower motor neuron disease of the spinal cord can develop. Death can occur, especially in children, within 3 to 5 days of CNS symptoms, or much later due to complications associated with hospitalization or cardiopulmonary status. A recent prospective study evaluated the clinical features and long-term prognosis of 118 children with encephalitis due to JEV in Malaysia.633 Only 44% of patients had full recovery, with 8% dying during the acute phase of the illness and 31% having persistent and severe neurologic sequelae. These included chronic seizures, motor dysfunction, and neuropsychiatric symptoms such as mental retardation and psychiatric disorders.
Pathologic Features of JEV Infection
JEV infection in the brain results in neuronal degeneration, necrosis, microglial nodule formation, and perivascular and parenchymal leukocyte infiltrates as well as focal hemorrhage. Parenchymal damage in the CNS is attributed to both direct cytopathic effect of the virus in nonrenewing populations of neurons and the resultant inflammatory state induced by activated microglia and infiltrating leukocytes. Although these histologic findings can occur throughout the brain, they usually are more restricted to the gray matter in the cortex, midbrain,
and brainstem, providing anatomic correlates for the tremor and dystonias associated with CNS infection. Focal lesions are seen predominantly in the thalamus and cerebral peduncles but also are commonly observed in the substantia nigra, cerebral and cerebellar cortices, and the anterior horn of the spinal cord,768 the latter of which is associated with a poliomyelitis-like acute flaccid paralysis.771 In patients who die rapidly, there may be little histologic evidence of inflammation, but instead, high levels of JEV antigen can be detected in morphologically intact neurons.362
and brainstem, providing anatomic correlates for the tremor and dystonias associated with CNS infection. Focal lesions are seen predominantly in the thalamus and cerebral peduncles but also are commonly observed in the substantia nigra, cerebral and cerebellar cortices, and the anterior horn of the spinal cord,768 the latter of which is associated with a poliomyelitis-like acute flaccid paralysis.771 In patients who die rapidly, there may be little histologic evidence of inflammation, but instead, high levels of JEV antigen can be detected in morphologically intact neurons.362
St. Louis Encephalitis Virus
History, Global Distribution, and Epidemic Cycle
St. Louis encephalitis virus (SLEV) is a mosquito-borne member of the JEV serocomplex capable of causing severe neurologic disease in humans. SLEV was first discovered in 1933 following a large epidemic of encephalitis in St. Louis, Missouri (1,095 cases and 225 deaths).185,508,675 More than 10,000 cases of severe illness and 1,000 deaths have since been attributed to SLEV infection, reflecting annual endemic transmission (∼50 cases/year) punctuated by epidemic periods that occur every 5 to 15 years.563 At least 41 epidemics of SLEV have occurred in the United States since 1933,185 the largest of these in 1975.167 During this epidemic, SLEV cases were reported in 29 states and the District of Columbia; the greatest number of illnesses occurred in Ohio, Mississippi, Indiana, and Illinois. Roughly 1,500 confirmed cases were reported, resulting in 171 fatalities. The most recent large outbreak of SLEV occurred in central Florida during 1990, resulting in 222 laboratory-confirmed cases and 14 deaths.543
SLEV is found in much of the New World; distribution ranges from Canada to Argentina, and across North America.675 SLEV is maintained in nature in enzootic cycles between Culex mosquitoes and passeriform and columbiform birds. Of interest, the transmission cycle of this virus varies by region due to differences in the biology of the primary vector mosquitoes.675 In the eastern and central United States, the principal vectors of SLEV are Culex pipiens and Culex quinquefasciatus mosquitoes. Culex tarsalis is the primary vector for SLEV in Western states, whereas Culex nigripalpus transmits SLEV in Florida. The avian hosts of SLEV in these transmission cycles include house finches, house sparrows, and mourning doves. The mechanism of virus transmission and amplification in South and Central America is less clear. SLEV has been isolated from 11 different mosquito genera, many of which feed primarily on mammals.
Both WNV and SLEV are antigenically related members of the JEV serogroup that share a similar transmission cycle between Culex mosquitoes and birds. How the introduction of WNV in North America has impacted the epidemiology of SLEV is of significant interest. Analysis of the number of neuroinvasive cases attributed to SLEV reported to the CDC between 1999 and 2007 revealed a threefold reduction by comparison with data in the pre-WNV era.674 Interpretation of this finding is complicated by changes in the intensity of surveillance and local testing for arboviral diseases in the years after the introduction of WNV. Because major epidemics of SLEV have occurred infrequently in the past, the modest number of clinical cases may simply reflect a nadir in the natural cycle of this virus. Alternatively, the existence of cross-reactive antibodies in WNV-immune avian reservoirs may disrupt the transmission cycle of SLEV via competition for avian hosts. Although the infection of house finches with WNV has been shown to confer protection from subsequent infection by SLEV, the reciprocal is not true. Prior exposure of finches to SLEV prevents mortality following WNV infection but not the low-level of viremia that is sufficient for transmission of WNV.232 Similar findings were reported in a golden hamster model of infection.817 The disappearance of SLEV from regions of California following introduction of WNV is consistent with the notion that competition may allow for the local displacement of the virus from historically endemic areas.676 Additional study and surveillance are required to clarify the dynamics and interactions between these two related pathogens in North America.
SLEV Diversity
Phylogenic studies grouped SLEV isolates into seven genetic lineages (I–VII), many of which were divided further into clades of related genotypes.421,538 These groups correspond roughly to the geographic distribution of each lineage of SLEV.830 For example, lineage I include viruses isolated in the western United States, whereas lineage V contains South American strains and an isolate from Trinidad. However, the relationship between phylogenic relatedness and geographic region is imperfect. SLEV strains vary considerably with respect to virulence in avian and mammalian hosts; these differences correlate roughly with geographic distribution.90,571 In addition to regional persistence, sequence analysis reveals that SLEV may be transported between regions.421
Clinical and Pathologic Features of SLEV Infection
As is the case for both WNV and JEV, the majority of SLEV infections of humans are clinically asymptomatic. The ratio of apparent to inapparent infections has been reported to range from 1:16 to 1:425.563 Increasing age is a significant factor influencing susceptibility to severe illness. Symptomatic illness is noted after an incubation period of 5 to 15 days and is characterized by mild malaise, fever, headache, nausea, myalgia, sore throat, and cough.103 Severe neurologic manifestations including encephalitis and aseptic meningitis may occur and can be fatal. Case fatality rates for SLEV range from 5% to 20%, with fatalities increasing in the elderly.675 Although most SLEV cases resolve spontaneously and without sequelae, many patients (30% to 50%) experience an extended convalescence lasting up to 3 years. This phase is characterized by headache, depression, memory loss, and weakness.103,675
Tick-Borne Encephalitis Viruses
History, Global Distribution, and Epidemic Cycle
Tick-borne encephalitis virus (TBEV) causes a fatal neurologic syndrome that primarily affects individuals ranging from northern China and Japan, through Russia, to parts of Northern Europe.525 TBEV infection was first described in 1931 after a pattern of seasonal meningoencephalitis cases in Austria was observed.732 In 1939, experiments confirmed that this seasonal encephalitis in humans was caused by a virus transmission by the tick, Ixodes persulcatus.923 Although a highly effective
formalin-inactivated vaccine has been implemented in some European countries (e.g., Austria) with marked reductions in case numbers,320 TBEV-induced morbidity and mortality continue to rise.799 Between 1990 and 2007, about 9,000 cases per year were reported in Europe and Russia799; currently, TBEV is believed to cause approximately 14,000 human cases per year, the majority of which occur in parts of Russia.277 This increase is thought to be due to changes in climate, population dynamics and range of permissive ticks, and shifts in land usage. Within Russia, Siberia has the highest number of TBEV cases, whereas the Czech Republic has the greatest incidence.525 The relative virulence of TBEV decreases with its westward spread, with the far-eastern subtype having a case-fatality rate of almost 40%.
formalin-inactivated vaccine has been implemented in some European countries (e.g., Austria) with marked reductions in case numbers,320 TBEV-induced morbidity and mortality continue to rise.799 Between 1990 and 2007, about 9,000 cases per year were reported in Europe and Russia799; currently, TBEV is believed to cause approximately 14,000 human cases per year, the majority of which occur in parts of Russia.277 This increase is thought to be due to changes in climate, population dynamics and range of permissive ticks, and shifts in land usage. Within Russia, Siberia has the highest number of TBEV cases, whereas the Czech Republic has the greatest incidence.525 The relative virulence of TBEV decreases with its westward spread, with the far-eastern subtype having a case-fatality rate of almost 40%.
 Figure 26.8. Life cycle of tick-borne encephalitis virus (TBEV). The transmission of tick-borne flaviviruses is connected to the life cycle of the vector due to a requirement for the tick to feed prior to transition through each of its developmental stage. Ticks are infected during this blood meal, molt, and then may infect a mammalian host. Nonviremic transmission between co-feeding ticks has also been shown to be an important mechanism of transmission and is not pictured. |
In the enzootic cycle, TBEV is maintained between ticks and different vertebrate hosts, with humans as incidental hosts (Fig. 26.8). TBEV is transmitted primarily by the hard tick Ixodes ricinus, although in Eastern Europe and Russia the principal vector is Ixodes persulcatus. Infection is seasonal, usually occurring between March and November,371 and coincides with seasonal peaks of feeding activity of the particular tick involved in transmission. TBEV is endemic from central Europe to Far East Asia, with cases reported in 34 countries.798 Ticks can become chronically infected after sampling viremic blood, or by transstadial or transovarial transmission. In addition, infected ticks can transmit virus to uninfected ticks during co-feeding on rodents.277,444 This is because the local skin environment supports TBEV replication, and migratory infected cells transport virus within the skin allowing for transmission in the absence of viremia.443
One exception to TBEV transmission by tick inoculation is the syndrome of biphasic milk fever, which results from oral infection and was first identified in Russia between 1947 and 1951. During milk fever epidemics, whole families contracted TBEV infection, and this was associated with the consumption of goat milk. Goats develop subclinical TBEV infection after tick bite and become the source of infectious virus after secretion into milk. Analogously, TBEV transmission to humans has been reported after consumption of unpasteurized cow or sheep milk or dairy products.277 These findings are supported by experiments in mice in which TBEV infection was established after oral feeding.654
TBEV Diversity
Based on sequence similarity, three main subtypes of TBEV exist: the Far Eastern genotype 1 (previously Russian Spring and Summer encephalitis), European genotype 2 (previously Central European encephalitis), and Siberian genotype 3 (previously west-Siberian). These TBEV genotypes are closely related218,339 and transmitted by the ticks Ixodes ricinus (European subtypes) and Ixodes persulcatus (Asian subtypes).277 Within these three genotypes, there is an approximately 1.2% to 1.7% difference at the amino acid level. The Far Eastern, European, and Siberian genotypes 1, 2, and 3 differ from each other by approximately 5% to 7% at the amino acid level. In addition to these three TBEV genotypes, two additional genotypes (4 and 5) have been described based on nucleotide and amino acid differences.193 Other viruses that are antigenically related across Europe, Asia, and North America are classified as part of the TBEV serocomplex,118 also termed the mammalian group of tick-borne flaviviruses. In addition to TBEV, this group includes Omsk hemorrhagic fever virus (OHFV), Louping ill virus (LIV), Langat virus (LGTV), Powassan virus (POWV), Kyasanur Forest disease virus (KFDV), Kadam
virus (KADV), Royal Farm virus (RFV), Gadgets Gully virus (GGYV), Alkhurma hemorrhagic fever virus (AHFV), and Karshi virus (KSIV). Of these viruses, TBEV, LIV, and POWV cause encephalitis in humans and animals, whereas OHFV, KFDV, and AHFV cause hemorrhagic fever.278,695 LGTV is a naturally occurring avirulent virus (analogous to Kunjin virus among WNV strains), and no clinical disease has been reported for KSIV, RFV, or GGYV.
virus (KADV), Royal Farm virus (RFV), Gadgets Gully virus (GGYV), Alkhurma hemorrhagic fever virus (AHFV), and Karshi virus (KSIV). Of these viruses, TBEV, LIV, and POWV cause encephalitis in humans and animals, whereas OHFV, KFDV, and AHFV cause hemorrhagic fever.278,695 LGTV is a naturally occurring avirulent virus (analogous to Kunjin virus among WNV strains), and no clinical disease has been reported for KSIV, RFV, or GGYV.
Clinical Features of TBEV Infection
About one-third of patients after inoculation with an infected tick will become symptomatic,371 with men affected twice as frequently as women, although this could reflect exposure bias. The incubation period for TBEV infection in humans varies, but for most individuals is approximately 1 to 2 weeks. A prodrome of fatigue, musculoskeletal pain, and headache lasts a few days, and is followed by an abrupt onset of fever, nausea, vomiting, and myalgias; this phase is associated with thrombocytopenia, leukopenia, and mildly elevated levels of liver enzymes in the serum. Subsequent to this, several clinical syndromes of TBEV infection develop, as reviewed previously277,371:
Febrile syndrome. This illness is characterized by high fever (39°C) with no evidence of neuroinvasion. It lasts from 1 to 5 days, and upon defervescence, patients recover completely.
Meningitis. This is the most common form of clinically apparent TBEV infection occurring in approximately 50% of individuals. After the onset of fever, symptoms worsen with progressive headache, nausea, vomiting, and photophobia. All patients exhibit a CSF leukocyte pleocytosis after lumbar puncture. Fever lasts 1 to 2 weeks, with gradual recovery.
Meningoencephalitis. This form occurs in approximately 10% of cases, is more severe, and is associated with damage to the CNS. Individuals become weak, lethargic, and develop focal signs of disease including hemiparesis, hemiplegia, seizures, and autonomic instability. Up to 30% of these cases are fatal, and survivors have long-term neurologic sequelae with slow convalescence.
Poliomyelitis-like disease. This is characterized by a prodrome of limb weakness or numbness that progresses to paralysis. Paralysis occurs more frequently in the upper limbs, with the proximal segments affected more often. Recovery is slow, partial, and occurs in only one-half of patients, with the remainder showing progressive deterioration.
Polyradiculitis. This syndrome has a biphasic course with fever, headache, and myalgia followed by defervescence. Approximately one week later the second phase starts and is characterized by pain and damage in peripheral nerves, sometimes coupled with meningitis. Recovery from this form of TBEV infection is usually complete.
Chronic or persistent infection. This form has been described in Siberia and Far East Russia, although not in Europe, and is believed to associate uniquely with the Siberian subtype of TBEV. Chronic or persistent infection is characterized by a late phase (months or even years later) deterioration of the neurologic sequelae that developed during the acute illness. Alternatively, chronic TBEV infection can begin with the acute phase of disease, such that neurologic symptoms occur years after a tick bite. Clinical symptoms can include epilepsy, Parkinsonian movement and cognitive disorders, and progressive muscle atrophy, ultimately with dementia and death ensuing. Although infectious virus has not been routinely recovered in autopsy studies, a TBEV strain was isolated from a patient who died of a progressive (2-year) form of tick-borne encephalitis 10 years after experiencing a tick bite.276
Postencephalitic syndrome. Both retrospective and prospective clinical trials have shown that TBEV infection is associated with a slow recovery period that has considerable long-term morbidity.289,370,554 This postencephalitic syndrome occurs in approximately 40% to 60% of patients, and includes memory disturbances, headache, and affective and gait disorders. The frequency of these symptoms was proportionately higher in more severe cases.
Pathologic Features of TBEV Infection
Gross pathologic analysis of the brain of humans who succumb to lethal TBEV infection shows edema and hyperemia. Microscopic lesions occur in a patchy distribution throughout the CNS but are most prominent in the brainstem, basal ganglia, thalamus, cerebellum, and spinal cord. The cerebral and spinal meninges show a diffuse leukocyte infiltration, predominantly with lymphocytes. In the parenchyma of the brain and spinal cord, perivascular infiltrates, microglial nodules, and necrosis of neurons is observed. Notably, Purkinje cell neurons in the cerebellum and anterior horn motor neurons in the spinal cord are preferentially targeted and injured by TBEV.371 Immunohistochemical analysis of brains from 28 autopsy cases250 showed prominent TBEV antigen staining in Purkinje cells, neurons of the dentate gyrus, the brainstem, and basal ganglia, with T lymphocytes detected in direct apposition to TBEV-infected neurons.
Pathogenesis and Immunity
Virus Entry and Tropism
Flavivirus entry into cells is mediated by the envelope proteins and can be considered in three relatively discrete steps (Fig. 26.9). The first step involves the attachment of the virus particle to the cell. Collisions between virions and target cells are not always productive. “Attachment factors” promote infection by increasing the duration of contact between the virion and cell surface, and thereby increase the likelihood that subsequent steps in the virus entry pathway will occur. Attachment factors are not strictly required for infection. In contrast, interactions with viral “receptors” promote required events during virus entry. Although the distinction between these two types of cellular factors is clear for some viruses (e.g., HIV), the cell biology of flavivirus entry remains poorly understood.
Several cellular factors have been suggested to function as attachment factors or receptors during the flavivirus entry (reviewed by Lindenbach, Murray, Thiel, and Rice in Chapter 25 of this volume). The interaction between flaviviruses and glycosaminoglycans (GAGs) have been documented.147,332,455,523 The binding site for these sulfated polysaccharides on the virion has been mapped to positively charged surfaces of the E protein.147,523 Passage of virus in cell culture selects for variants that bind more efficiently to GAGs, although this adaptation appears to be associated with reduced fitness in vivo.454,456,523 Treatment
of cells with heparan sulfate can inhibit infection.147,457,485 GAGs are thought to promote more efficient attachment to cells via electrostatic interactions with the virus particle.
of cells with heparan sulfate can inhibit infection.147,457,485 GAGs are thought to promote more efficient attachment to cells via electrostatic interactions with the virus particle.
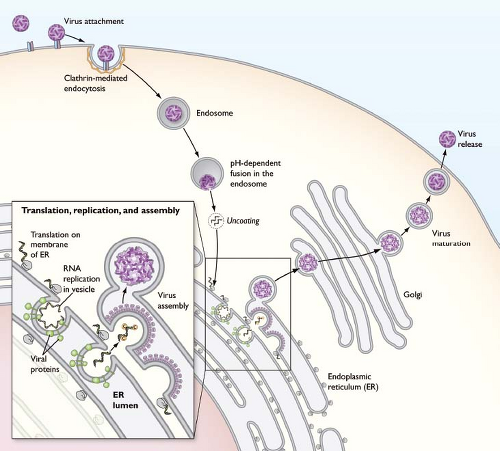 Figure 26.9. The replication cycle of flaviviruses. Flaviviruses bind cells of the host through poorly defined interactions with one or more molecules on target cells. Viruses are internalized via clathrin-mediated endocytosis and fuse with membranes of the late endosome in a pH-dependent manner. Viral RNA replication begins shortly thereafter in association with membranes of the host cell. Cells actively replicating flaviviruses reveal striking host membrane rearrangements thought to coordinate the processes of genomic RNA replication and virus assembly. Virus particles assemble at and bud into the endoplasmic reticulum and are secreted from the cell. During egress, virion maturation occurs in the acidic compartments of the Golgi and is characterized by cleavage of the prM protein by a furin-like protease. |
Cellular lectins also increase the efficiency of flavivirus attachment. CD209 Dendritic Cell-Specific Intercellular Adhesion Molecule-3-Grabbing Non-integrin (DC-SIGN) is a calcium-dependent c-type lectin that serves as an attachment factor for several classes of viruses (reviewed in839), including some flaviviruses.183,605,811 These interactions are mediated by N-linked sugars on the prM and E proteins of the virion.182,406,480,504 CD209 is expressed in vivo on a subset of dendritic cells (DCs) and macrophages.839 The infectivity of DCs by DENV correlates with CD209 expression; immature DCs express CD209 and are more permissive to infection than mature DCs expressing lower levels of CD209.811,893 Antibodies against CD209 or soluble forms of this lectin are capable of blocking DENV infection of DCs.605,811 Experiments with truncated forms of CD209 suggest that internalization of CD209 is not required to increase the efficiency of virus attachment to selected cell types.503 CD209L Dendritic Cell-Specific Intercellular Adhesion Molecule-3-Grabbing Non-integrin-Related protein (DC-SIGNR)183,811 and the mannose
receptor (MR)556 have also been identified as attachment factors for flaviviruses.
receptor (MR)556 have also been identified as attachment factors for flaviviruses.
Recent studies have identified members of the TIM and TAM families of phosphatidylserine receptors that function as attachment and potentially signaling factors for flaviviruses. These cellular protein directly (in the case of TIM1) or indirectly (in the case of TAM proteins) bind lipids incorporated into the membrane of the virus particle. The expression pattern of this family of molecules may explain, in part, the broad cellular tropism of these viruses in vitro. Furthermore, the interaction of cellular proteins with the lipid envelope of the virion, thought to be buried by the dense icosahedral array of E proteins described above), raises questions about the structure(s) of infectious flaviviruses.543a
Flavivirus enter cells via clathrin-mediated endocytosis.2,152,262,424,838 Elegant single-particle tracking studies of DENV suggest that virions move across the surface of cells until they encounter preformed clathrin-coated pits. Virus particles are then internalized and traffic into late Rab7-positive endosomal compartments where viral fusion occurs.838 Fusion between viral and cellular membranes is triggered by the acidic environment of endosomes. How viruses sense the low-pH environment is not completely understood, but may involve the protonation of key histidine residues on the E protein.241,375,608 Fusion may also be governed by the lipid composition of the endosome.909 That flaviviruses have the ability to fuse with synthetic liposomes devoid of proteins indicates that this process does not require interactions with a cellular receptor.162,264
The molecular basis for the tropism of flaviviruses is unknown. A wide variety of cell lines representing different lineages and species may be infected in vitro. This suggests cellular factors involved in virus entry are either highly conserved (from mosquitoes to man) or redundant. Targets for flavivirus infection in vivo appear more restricted and include monocytes, macrophages, hepatocytes, neurons, endothelial cells, and DCs.38,704 Tropism may be regulated at a postentry level through the activities of interferon and interferon-stimulated genes.704,859
Mechanisms of Dissemination
Blood-Borne Viruses
For both viscerotropic (e.g., DENV and YFV) and encephalitic (e.g., WNV, JEV, and TBEV) flaviviruses, the skin is the likely initial infection site after insect inoculation, with resident dendritic cells117,529 or epidermal keratinocytes478 believed to be the primary target cells. The dose of virus inoculated by the mosquito under conditions of natural infection is not known precisely, but likely ranges from 103 to 105 plaque-forming units (PFU),731,791 depending on the flavivirus and insect vector. Active WNV replication can be detected at the subcutaneous site of infection within one day of infection,107 and virus spread to the lymph node occurs in animals infected by mosquitoes or with mosquito salivary extracts.731,792 Proteins in mosquito saliva alter cytokine levels and other components of innate immunity, leading to local immunosuppression or dysregulation,729 and enhanced spread and replication.
Flaviviruses disseminate to local lymph nodes either associated with migratory infected dendritic cells663 or as free virus that transudates directly into lymphatic fluid.369 Macrophages on the floor of the subcapsular sinus and in the medulla of lymph nodes capture viral particles efficiently, serving as possible targets of virus amplification infection and initiators of innate and adaptive immune responses.369 Virus produced in the draining lymph nodes likely spreads to intravascular venous compartments via efferent lymphatic drainage. Virus in the bloodstream can directly infect blood cells or visceral tissues, which can result in further dissemination and secondary viremia.
The infectivity of flaviviruses in plasma, the fluid component of blood, appears remarkably short, with a half-life in mice ranging from 2 to 10 minutes for DENV and WNV, respectively.242 The loss of infectivity is due in part, to complement (C3 and C4 components) opsonization via mannose-binding lectin recognition of N-linked glycans on the surface of virions.242 The short half-life of infectious flavivirus in plasma may also reflect sequestration and removal by different visceral organs.355 Alternatively, flaviviruses may transit rapidly into the cellular compartment of blood. One study of patients with DENV infections of different disease severity showed DENV antigen (prM and NS3) predominantly in cells of monocyte (CD14+, CD32+) lineage, with up to 80% to 90% of cells of expressing viral antigen.216 This finding of DENV in blood monocytes is consistent with prior literature526 but contrasts with newer studies in rhesus macaques, suggesting that platelets become positive for dengue antigen during the course of infection.632 Finally, another explanation for the rapid drop of plasma infectivity is that flaviviruses adhere readily to erythrocytes in whole blood.683
Neurotropic Viruses
Flavivirus neuropathogenesis requires neuroinvasiveness, the capacity to enter the CNS, and neurovirulence, the ability to propagate efficiently within cells of the CNS. In classical studies, phenotypic distinctions were made among different arthropod-borne viruses on the basis of replication efficiency and pathogenic potential in peripheral versus CNS tissues.5 A main principle was the relationship between peripheral virus burden and the propensity to cause neuroinvasion. Viruses with a low capacity to replicate in the periphery generally had less neuroinvasive potential, regardless of their intrinsic neurovirulence. Aerosol-acquired and, perhaps, mucosal infections are possible exceptions, as these may use alternate routes of CNS entry.
Data from several studies indicate that the time of onset, magnitude, and duration of viremia, as well as the integrity of the host immune system influences the risk of entry into the CNS. Therefore, the neuropathogenic potential of most flaviviruses is a balance between the replication efficiency and the effectiveness of early host defenses in clearing viremia. Neuroinvasiveness is affected by both viral and host factors. Based on genetic analysis of virulent and attenuated strains of JEV, TBEV, YFV, and WNV, viral determinants of neuroinvasiveness map primarily to the E protein.54,56,130,541,615,616 The mechanisms associated with these genetic determinants have not been determined, but are believed to relate to increased viral infectivity of key target cells through enhanced binding and penetration.
Animal models of infection of encephalitic flaviviruses have begun to define factors that govern virus entry into the brain and spinal cord. Crossing of the blood–brain barrier (BBB) likely occurs through a hematogenous route, as increased viral burden in the serum correlates with earlier and enhanced viral entry into the brain.204 Accordingly, changes in endothelial cell permeability may facilitate CNS entry; these may be triggered by vasoactive cytokines509,868 or activation of matrix metalloproteinases that degrade the BBB extracellular matrix.845,864 Additional possible mechanisms may contribute to CNS infection of flaviviruses, including the following: (a)
direct infection or passive transport through the endothelium,212,488,846 and (b) infection of olfactory neurons and rostral spread from the olfactory bulb.107 Access through the olfactory bulb is believed to occur either after infection by the aerosol or intranasal route602,618,669 or in the context of hematogenous dissemination of virus.542 The olfactory bulb is vulnerable to direct infection because of the exposure of its nerve terminals within the olfactory mucosa; (c) a “Trojan horse” mechanism in which virus is transported by infected immune cells that traffic to the CNS865; (d) access to the CNS after breakdown of BBB integrity139,412; and (e) direct axonal retrograde transport from infected peripheral neurons.351,584,705,860 Although much has been learned from infection studies in mice and hamsters, the precise mechanisms of CNS entry of encephalitic flaviviruses in humans and other animals requires additional study.
direct infection or passive transport through the endothelium,212,488,846 and (b) infection of olfactory neurons and rostral spread from the olfactory bulb.107 Access through the olfactory bulb is believed to occur either after infection by the aerosol or intranasal route602,618,669 or in the context of hematogenous dissemination of virus.542 The olfactory bulb is vulnerable to direct infection because of the exposure of its nerve terminals within the olfactory mucosa; (c) a “Trojan horse” mechanism in which virus is transported by infected immune cells that traffic to the CNS865; (d) access to the CNS after breakdown of BBB integrity139,412; and (e) direct axonal retrograde transport from infected peripheral neurons.351,584,705,860 Although much has been learned from infection studies in mice and hamsters, the precise mechanisms of CNS entry of encephalitic flaviviruses in humans and other animals requires additional study.
Mechanisms of Immune Control: Innate Immunity
Cellular Innate Immunity
Macrophages
Although only limited studies have directly addressed the function of cellular innate immunity in flavivirus infection, emerging data suggest that macrophages play key roles in orchestrating control of infection. Macrophages can limit infection though direct viral clearance, enhanced antigen presentation to B and T cells, and production of proinflammatory or antiviral cytokines and chemokines.426,527 The protective role of macrophages is highlighted by studies in mice, which demonstrated exacerbated WNV, TBEV, DENV, or YFV disease after selective macrophage depletion.63,234,390,663,924 Macrophages may control flaviviruses through the production of nitric oxide (NO) and other reactive oxygen intermediates after stimulation of inducible nitric oxide synthetase (NOS-2).422,484,712,713 Activation of macrophages in response to flavivirus infection also promotes release of type I interferon (IFN), tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-8, and other cytokines, some of which have antiviral activity and reduce viral replication, at least in culture.757 Despite their protective role in innate defense, macrophages also are targets of infection by some flaviviruses416,441,684 and have the potential to contribute to pathogenesis through antibody-dependent enhancement of infection mediated by Fc-γ and complement receptors.123,261,641 The macrophage cell surface receptor CLEC5a independently has been reported to interact with DENV directly, resulting in DAP12 phosphorylation and the release of proinflammatory cytokines.144 Therefore, in some circumstances macrophages can contribute to flavivirus-induced disease, although the contribution to clearance versus pathogenesis may vary depending on the specific virus, the presence of preexisting nonneutralizing antibodies, and the specific proinflammatory molecules that are produced.
Neutrophils
Although polymorphonuclear leukocytes (neutrophils) are among the first circulating leukocytes to respond to infection or inflammatory stimuli, their function in flavivirus infection remains uncertain. Some studies suggest a protective function; however, others indicate that neutrophils can contribute to flavivirus pathogenesis. A protective role was reported in the context of WNV infection as macrophages produced neutrophil chemoattractive chemokines (CXCL1 and CXCL2), neutrophils rapidly migrated to the site of infection, and mice depleted of neutrophils 1 or 2 days after virus infection developed higher viremia and experienced earlier death.30 Paradoxically, if neutrophils were depleted prior to infection, viremia was reduced and survival was enhanced.30 Analogously, depletion of neutrophils resulted in prolonged survival and decreased mortality in Murray Valley encephalitis virus-infected mice, and neutrophil infiltration and disease correlated with NOS-2 expression within the CNS.14 Finally, transcriptional gene signatures from whole blood showed a greater abundance of neutrophil transcripts in patients who progressed to DSS, a finding supported by higher plasma levels of proteins associated with neutrophil degranulation.334 Although further studies are warranted, neutrophils may prevent or promote flavivirus disease, depending on the specific virus and immunologic context.
Dendritic Cells
Human peripheral blood contains two types of dendritic cells (DCs), plasmacytoid DCs (pDCs) and myeloid DCs (mDCs), which can be distinguished based on function and distinctive surface markers. pDCs lack phagocytic capacity and are less efficient in capturing and presenting antigens to T cells, but they produce extraordinarily high levels of type I IFN in the presence of viruses or bacteria,759 and are thus considered to play a crucial role in antiviral immunity.802,803 Low levels of DENV replication were observed in pDCs, but proinflammatory cytokines were produced rapidly and could accumulate to high levels. This cytokine response was not dependent on viral replication, but dependent on endosomal toll-like receptor 7 (TLR7), and could be induced by purified DENV RNA.797,862 In prospective clinical studies, the absolute number of circulating pDCs remained stable early in moderately ill children with dengue fever or other nondengue, febrile illnesses. However, there was an early decrease in circulating pDCs in children who subsequently developed DHF, as a blunted blood pDC response was associated with an altered innate immune response, higher viremia levels, and severe disease.646 Of interest, the host origin of the flavivirus influences the response that is generated by pDCs, as WNV grown in mammalian cells was a potent inducer of IFN-α secretion in pDCs, whereas pDCs failed to produce IFN-α when exposed to WNV grown in mosquito cells.762
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


