TRAUMA
The eye is excised after severe ocular trauma if no potential for visual recovery exists. Blunt trauma to the eye may rupture the eyeball, especially at the junction of the cornea and sclera or immediately posterior to the insertion of the rectus muscles where the sclera is thinnest. Potential complications of ocular trauma include blood within the anterior chamber (hyphema) with associated corneal discoloration that is caused by hemoglobin deposition (corneal blood staining) and separation of the ciliary body from the iris (iridodialysis) or sclera (cyclodialysis) as well as cataracts, retinal detachments, and choroidal rupture.
FOREIGN BODIES
Foreign bodies commonly enter the eye as projectiles or as fragments accompanying branches or other sharp objects that perforate the globe. Vegetable matter, hair, and skin may enter the intraocular tissues after an explosive or perforating injury. These agents incite an inflammatory reaction that occasionally may be granulomatous in nature.
Some sterile foreign objects do not incite intraocular inflammation or cause specific adverse effects, whereas others cause a significant tissue response. Intraocular foreign bodies containing iron are particularly toxic to the retina, and they may cause a diffuse deposition of iron throughout the eye (13). Copper-rich foreign bodies incite a significant intraocular acute inflammatory reaction (14). Other metals such as lead, zinc, nickel, aluminum, and mercury may also evoke intraocular inflammation.
INFLAMMATION
Inflammation of the eye may involve the intraocular contents but spare the sclera and cornea (endophthalmitis), or it may affect the cornea and sclera in addition to the ocular contents (panophthalmitis). Both endophthalmitis and panophthalmitis may follow ocular trauma, surgery, or the hematogenous spread of a systemic infection. The distinction between endophthalmitis and panophthalmitis is clinically important because infections causing panophthalmitis potentially expose the patient’s orbit to microorganisms, whereas in infectious endophthalmitis, the cornea and sclera encase the intraocular infection, similar to an encapsulated abscess.
In both endophthalmitis and panophthalmitis, a profuse polymorphonuclear leukocytic infiltration is present, and intraocular hemorrhage may also be seen. The involved intraocular tissues are usually necrotic and disorganized, and the causal bacteria or fungi may be identified with special stains. A mild endophthalmitis due to Propionibacterium acnes sometimes follows cataract extraction (15,16). It has a minimal inflammatory reaction. A wide variety of organisms can cause intraocular inflammation (15–21).
Aside from sympathetic uveitis, granulomatous inflammation of the eye occurs in some conditions (Table 24.2). An important granulomatous endophthalmitis occurs around the lens as part of an immunologic reaction to lens proteins (phacoanaphylactic endophthalmitis) (22,23).

PHTHISIS BULBI
Numerous pathologic processes eventually culminate in an atrophic disorganized eye. Because such eyes with phthisis bulbi almost always contain significant amounts of lamellar bone, decalcification is usually required before the globe can be cut and submitted for tissue processing. The intraocular ossification can be detected on radiographic examination of the enucleated eye (24). The bone usually contains marrow with adipose tissue and blood vessels and occasionally megakaryocytes as well as erythrocytic and myelocytic lineage precursors (25). Phthisical eyes usually manifest extensive scleral thickening, chronic retinal detachment, and the intraocular contents are markedly disorganized. A fibrous diaphragm often extends circumferentially from the ciliary body behind the lens (cyclitic membrane). As a rule, a histologic examination of phthisical eyes fails to disclose evidence of the initial condition that led to this condition. Rarely, phthisical eyes have contained an unsuspected intraocular melanoma, lymphoma, or adenocarcinoma (26,27).
GLAUCOMA
The term glaucoma refers to a group of disorders that develop an optic neuropathy that is accompanied by a distinct excavation of the optic nerve head and an incremental loss of visual field sensitivity. In most cases, the intraocular pressure is increased, and, most notably, this damages the retina and optic nerve. Several types of glaucoma are recognized and are classified as follows: primary glaucoma, which is not associated with significant antecedent ocular disease, and secondary glaucoma, which is due to some ocular pathologic process. Primary glaucoma can result from a blockage of the drainage of the aqueous humor distal to the anterior chamber angle (primary open-angle glaucoma) and from a narrow anterior chamber angle (primary narrow-angle glaucoma) (28). Increased intraocular pressure, abnormal visual fields, and optic nerve damage are secondary effects of glaucoma.
Some surgical procedures used in the treatment of glaucoma result in excised tissue specimens that are submitted for pathologic evaluation at some institutions. For example, a small fragment of the trabecular meshwork is often excised (trabeculectomy) to enhance the drainage of aqueous humor from the eye to decrease intraocular pressure. This procedure produces a minute fragment of tissue that is often less than 1 mm in diameter. The processing of such specimens is not clinically important, but communication between the surgical pathologist and the histotechnician responsible for embedding such specimens is essential for proper tissue orientation; the use of a dissecting microscope is required. Light or transmission electron microscopic examination of trabeculectomy specimens is often unrewarding, but the trabecular meshwork, melanin pigment, Schlemm canal, ciliary muscle, or the peripheral cornea may be disclosed (29).
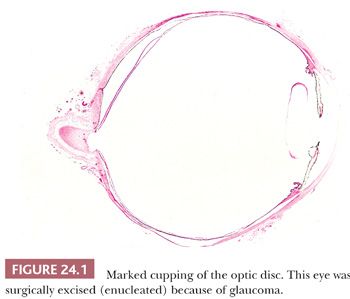
In end-stage glaucoma, the painful blind eyes may require enucleation. Morphologic evidence of increased intraocular pressure includes atrophy of the nerve fiber and ganglion cell layer and excavation of the optic nerve disc (glaucomatous cupping of the optic disc) (Fig. 24.1). A peculiar degeneration of an atrophic optic nerve is characterized by an accumulation of hyaluronic acid that stains positively with the Hale colloidal iron technique or the Alcian blue stain (Schnabel cavernous atrophy). An examination of a glaucomatous eye may also disclose the cause of congenital or secondary glaucoma. For example, fibrocollagenous adhesions may be present between the posterior surface of the peripheral cornea and the iris (peripheral anterior synechiae), and this may have obstructed the aqueous humor outflow.
CORNEA
EVALUATION OF CORNEAL TISSUE
Most surgically excised corneal specimens represent tissue obtained at the time of a full-thickness corneal transplant (penetrating keratoplasty). However, deep anterior lamellar keratoplasty (a partial-thickness corneal graft) has been increasing over the past years (30). Corneal biopsy specimens (31) are also sometimes performed and submitted for histopathologic evaluation. During corneal grafts, tissue is surgically removed from the cornea with the aid of a trephine so that a button approximately 8 mm in diameter is obtained. The manner by which corneal tissue is processed depends on the suspected pathologic process. Most specimens are fixed in formalin and processed for light microscopy according to standard procedures. Certain specimens, however, need special handling to ensure that the correct diagnosis is made. For example, some corneal disorders, such as Schnyder corneal dystrophy and other lipid keratopathies, need special fixatives or frozen sections to preserve the abnormal accumulations. Meesmann, Thiel-Behnke, and Reis-Bücklers corneal dystrophy require transmission electron microscopy for a morphologic diagnosis, but these dystrophies can now be diagnosed with molecular genetic techniques by using DNA from blood, tissue, or buccal swabs (32,33).
For histopathologic evaluation, a representative cross section through the center of each corneal button that includes any opacification or other abnormalities should usually be processed. Because the normal monolayer of corneal endothelium at the back of the cornea is easily disrupted, corneal specimens should be handled with care and should be cut with a sharp razor so that the lesion of interest is present within the portion of cornea submitted for microscopic examination.
CORNEAL GRAFTS
Indications for Corneal Grafts
Frequent indications for penetrating keratoplasty include keratoconus; failed corneal grafts; corneal endothelial decompensation (bullous keratopathy); Fuchs endothelial dystrophy certain corneal dystrophies; chronic keratitis, which is most often caused by herpes simplex; and corneal scarring resulting from acute nonspecific keratitis, trauma, etc. (30,34,35).
Graft Failure
Most currently performed corneal transplants are successful, and they provide long-term improvements in visual acuity for individuals with certain corneal diseases. In contrast to grafts of most tissues or organs, those involving the cornea are usually successful without compatibility matching of donor and recipient tissues (36). Some fail for various reasons, including endothelial decompensation (discussed under “Bullous Keratopathy”) (30), recurrent disease (37–40), immunologic graft rejection (41–44), or improper surgical technique. Epithelial irregularities, stromal vascularization, endothelial cell loss, and retrocorneal fibrous membranes often characterize failed corneal grafts, but a pronounced stromal inflammatory infiltrate is rarely evident. Unlike Fuchs endothelial corneal dystrophy, the basic abnormality of the macular, lattice, granular, and the other corneal dystrophies may recur in the corneal grafts (37–40,45).
NONSPECIFIC RESPONSES
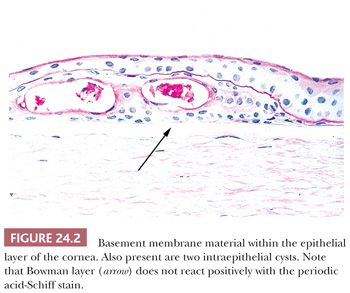
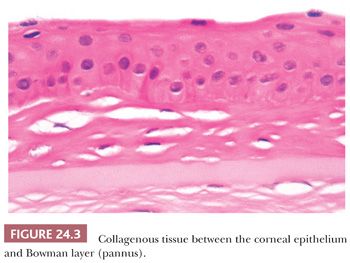
Different pathologic processes cause nonspecific histopathologic responses, and these may be evident in the corneal tissue. For example, intraepithelial vesicles and bullae between the epithelium and Bowman layer may follow corneal edema, especially of the epithelium (Fig. 24.2). An aberrant basal lamina develops within the corneal epithelium during the healing of some injuries (Fig. 24.2). Fibrous tissue, sometimes with blood vessels and mononuclear inflammatory cells, may accumulate between the epithelium and Bowman layer (pannus) (Fig. 24.3).
Blood vessels are present in the superficial or deep stroma of the normally avascular cornea in numerous pathologic states associated with inflammation. With aging, corneal endothelial cells diminish in number, and Descemet membrane thickens. A diffuse, irregular thickening of Descemet membrane accompanies some long-standing degenerative changes of the endothelial layer. Fibrous retrocorneal membranes between the Descemet membrane and the corneal endothelium may follow grafts or various inflammatory processes of the cornea (46).
CALCIFIC BAND KERATOPATHY
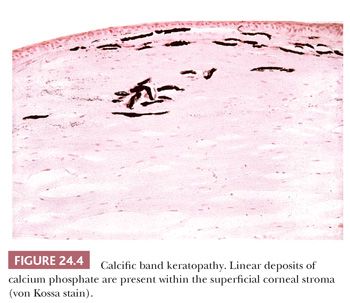
Under a variety of circumstances, calcium is deposited in the cornea (47–50), particularly in the Bowman layer and the superficial corneal stroma (calcific band keratopathy) (Fig. 24.4). In its early stages, a faint basophilic stippling of Bowman layer typifies calcific band keratopathy, and, in advanced cases, the entire thickness of the Bowman layer is involved.
CHRONIC ACTINIC KERATOPATHY (CLIMATIC DROPLET KERATOPATHY)
Amorphous globules of protein accumulate in the superficial stroma of the interpalpebral portion of the cornea in an entity known by numerous terms, including chronic actinic keratopathy (51), climatic droplet keratopathy (52), and spheroidal degeneration (53). The condition, which initially involves the periphery of the cornea, varies in severity and increases in incidence and intensity with age. It is particularly pronounced in individuals exposed over long periods to excessive ultraviolet light. Similar globules can accumulate nonspecifically in corneas with various underlying disorders (“spheroidal degeneration”).
BULLOUS KERATOPATHY
The corneal endothelium helps to maintain proper hydration of the cornea. Meaningful injury to this monolayer of cells, which does not regenerate significantly in humans, may result in corneal epithelial and stromal edema, subepithelial bullae, and decreased visual acuity. The many causes of this so-called bullous keratopathy include immunologic rejection of the corneal endothelium, and Fuchs corneal dystrophy. Bullous keratopathy also may be precipitated by cataract extraction (aphakic bullous keratopathy), sometimes after the combined implantation of a prosthetic intraocular lens (pseudophakic bullous keratopathy) (54). Descemet-stripping endothelial keratoplasty (DSEK) is a technique currently being performed in eyes with corneal endothelial decompensation and failed penetrating keratoplasty (55,56).
Corneal buttons removed from patients with bullous keratopathy manifest intraepithelial vesicles, bullae between the epithelium and the Bowman layer, and markedly fewer endothelial cells than normal. An intraepithelial basement membrane and a mild subepithelial pannus may also be present. Mild degrees of stromal edema are difficult to discern histologically, but increased corneal thickness in association with an absence of the normal artifactual clefts between the collagen lamellae in routinely processed tissue sections is suggestive of corneal edema. Neovascularization and inflammation can be observed in older lesions (57).
KERATOCONUS
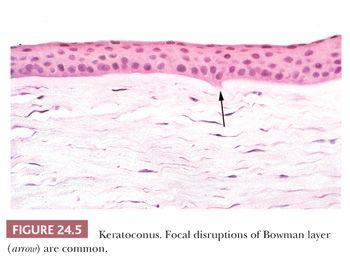
Noninflammatory thinning of the central corneal stroma takes place in keratoconus, causing a cone-shaped cornea (58). Scarring and astigmatism associated with this disorder often prevent adequate refractive correction and decrease visual acuity. A brown, stainable intraepithelial iron arc or ring frequently surrounds the conical portion of the cornea (Fleischer ring). Numerous breaks in the Bowman layer that are associated with the thinning of the central corneal stroma characterize advanced cases (59) (Fig. 24.5). The endothelium in corneas with keratoconus is usually unremarkable, but endothelial cell loss may accompany ruptures of Descemet membrane (“corneal hydrops”). Rarely, keratoconus recurs in the graft (60).
CORNEAL DYSTROPHIES
The corneal dystrophies (CDs) are a heterogeneous group of inherited bilateral corneal disorders (Table 24.3). Clinically, CD can be divided into three groups: anterior CD (affects primarily the corneal epithelium and Bowman layer and superficial corneal stroma), stromal CD (affects corneal stroma), and posterior CD (affects Descemet membrane and the corneal endothelium). The prevalence of the different CD varies in different countries and even within the different parts of some countries. Fuchs endothelial dystrophy accounts for most CD specimens submitted for pathologic examination in the United States. Other corneal specimens that reach the pathology laboratory have macular, lattice, or granular CD. Many of these diseases have been mapped to specific chromosomes, and the genes have been identified in many of them. Different genetic mutations have been related with CDs development. Most frequently involved genes are TGFBI, CHST6, KRT3, KRT12, PIP 5k3, SLC 4AIII, TATICSt2, and UBIADI.

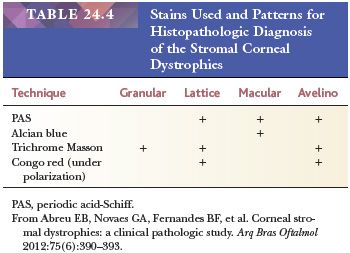
In fact, the recent availability of genetic analyses has demonstrated the shortcomings of the current phenotypic method of CD classification. It has been demonstrated that abnormalities in different genes can cause a single phenotype, whereas different defects in a single gene can cause different phenotypes. Moreover, some disorders termed corneal dystrophy do not appear to have a genetic basis (61,62). Recently, classification of the CDs based on the genetic changes has been proposed (63). Space restrictions prevent a description of all CDs. For details, the reader is referred elsewhere (33,34). A table with some common special stains to evaluate stromal CD is displayed (Table 24.4) (64).
Fuchs Corneal Dystrophy
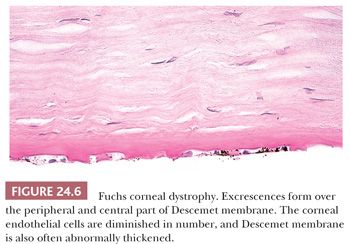
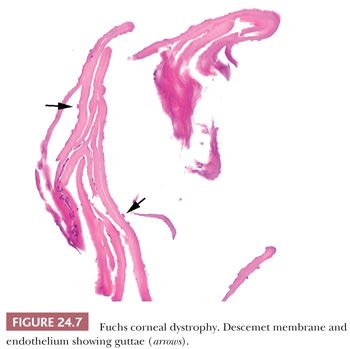
Fuchs CD is characterized by the presence of multiple wartlike excrescences on the Descemet membrane (corneal guttae) (Fig. 24.6) and the histologic features of bullous keratopathy. The centrally located corneal guttae are morphologically identical with the structures that form in the peripheral cornea with normal aging (Hassall-Henle bodies). However, Hassall-Henle bodies are too peripheral in location to be observed in specimens obtained during a routine penetrating keratoplasty. In some cases, the pathologist receives just the Descemet membrane and endothelium showing the guttae (Fig. 24.7). Corneal guttae are not specific for Fuchs CD; they are also found in macular CD (described under “Macular Corneal Dystrophy”) and in some cases of interstitial keratitis and keratoconus. The presence of inconspicuous ghost vessels in the most posterior corneal stroma distinguishes interstitial keratitis from Fuchs CD. Alterations in the gene COL8A2 have been recently linked to Fuchs CD (65).
Macular Corneal Dystrophy
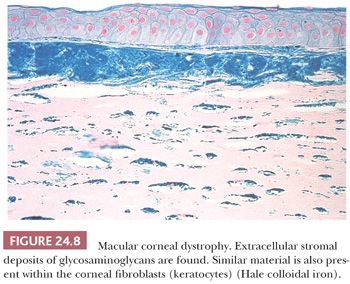
Corneas from patients with macular CD are characterized by an accumulation of a keratan sulfate–related glycosaminoglycan within both the fibroblasts and the endothelium of the cornea as well as among the collagen lamellae and in the Descemet membrane. The Hale colloidal iron technique and the Alcian blue stain are particularly useful in coloring the abnormal accumulations (66) that result from a mutation in the CHST6 gene on human chromosome 16 (16q22.1) (32) (Fig. 24.8).
Corneal Dystrophies with Amyloid Deposition
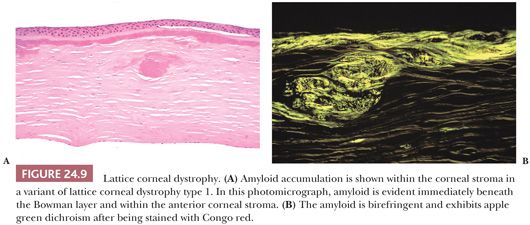
The inherited CDs with amyloid deposition are characterized by irregular linear opacities resulting from stromal amyloid deposition (Fig. 24.9) in corneas with an unremarkable Descemet membrane and endothelium. Amyloid is apparently localized to the cornea in most cases of lattice CD, but, in one type (lattice CD type 2), it is a manifestation of a systemic disease (familial amyloid polyneuropathy type 3, Finnish or Meretoja type). Like deposits of amyloid elsewhere, the corneal deposits in the lattice CDs react positively with the Congo red stain and with other methods for amyloid. Lattice CD type 1 is caused by specific mutations in the TGFBI gene, and affected individuals often have recurrent epithelial erosions and subepithelial amyloid or collagenous plaques. The amyloid in this dystrophy reacts with antibodies to the transforming growth factor-β–induced protein (67).
Amyloid also accumulates in the corneal stroma in numerous nonspecific, long-standing ocular disorders, including trauma, keratoconus, trachoma, uveitis, the retinopathy of prematurity, phlyctenular keratoconjunctivitis, sympathetic ophthalmia, and glaucoma (68). In most of the corneal amyloidoses, the nature of the amyloid remains unknown, but, in lattice CD type 1, mutant transforming growth factor-β–induced protein is a major component (67). In lattice CD type 2, the amyloid is derived from a part of mutant gelsolin (69,70). A significant amount of lactoferrin accumulates within the cornea in gelatinous droplike dystrophy of the cornea (familial subepithelial corneal amyloidosis) (68), an inherited corneal disorder due to a mutation in the TACSTD2 (formerly known as M1S1) gene on human chromosome 1 (1p32) (32).
Granular Corneal Dystrophy
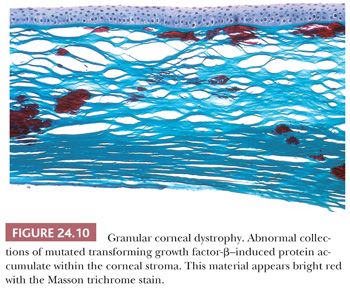
In granular CD, abnormal subepithelial and anterior stromal deposits appear bright red with the Masson trichrome stain (Fig. 24.10). The posterior stroma, Descemet membrane, and corneal endothelium are usually unaffected. This dystrophy results from a mutation in the TGFBI (BIGH3) gene (32,66), and the corneal deposits react with antibodies to transforming growth factor-β–induced protein (71). The TGFBI mutation responsible for granular CD type 1 is usually R55W and it differs from those causing granular CD type 2 and type 3 (Reis-Bücklers CD) (33,34).
KERATITIS CAUSED BY ORGANISMS
Bacteria, fungi, viruses, or protozoa (72–76) (Table 24.5) frequently infect the cornea, and, especially if corneal perforation is imminent, corneal transplantation may be performed on such tissue. Corneal biopsies are occasionally used to identify the offending organism in acute keratitis (31). Those who wear contact lenses are particularly susceptible to keratitis from Pseudomonas species (77) and Acanthamoeba (78,79). Keratitis caused by the microfilaria of the nematode Onchocerca volvulus is a leading cause of blindness worldwide, but affected individuals are rarely treated in the United States (80).

Although the pathogenic agent influences the nature of the tissue reaction in acute ulcerative keratitis, the histopathologic features are strikingly similar in most instances; these include destruction of the corneal epithelium, Bowman layer, and stroma, as well as necrosis and a prominent polymorphonuclear leukocytic infiltrate. With corneal perforation, discontinuities of Descemet membrane develop, and inflammatory debris adheres to the posterior surface of the cornea. The causative microorganism is often difficult to detect in tissue sections without the aid of special stains. Colonies of some bacteria, such as Streptococcus viridans, may produce crystalline-like stromal opacities in the absence of an inflammatory cell infiltrate (“infectious pseudocrystalline keratopathy”) (81).
Acanthamoeba
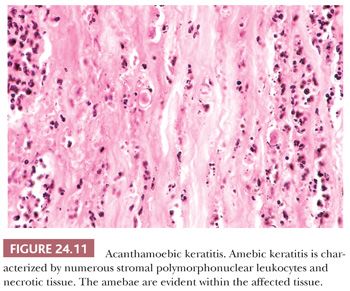
Keratitis caused by Acanthamoeba (A. castellani, A. culbertsoni, A. polyphaga, or A. rhysodes) is a well-recognized complication in those who wear contact lenses. This protozoan can be recognized in hematoxylin and eosin (H&E)–stained sections, but special stains (calcofluor white, periodic acid-Schiff, methenamine silver, Giemsa), immunofluorescent techniques, and transmission electron microscopy have been advocated for the diagnosis of amebic keratitis (Fig. 24.11). Amebic cysts and trophozoites are most often found near areas of stromal necrosis (82,83). In the absence of specific immunohistochemical (IHC) methods, the differentiation of trophozoites from reactive corneal fibroblasts may be difficult.
Herpes Simplex
Human herpesvirus-1 (herpes simplex type 1) is the most common viral cause of clinically significant corneal disease (84). The histopathologic features of long-standing keratitis resulting from this virus are nonspecific. The epithelium may be irregular in thickness. Bowman layer is frequently disrupted, and pannus formation is often present. Neovascularization and an infiltrate of mononuclear inflammatory cells that is composed primarily of lymphocytes may be present in the corneal stroma. Rarely, a granulomatous infiltrate is present in the deep stroma or surrounding Descemet membrane. Granulomatous keratitis is not specific for herpes simplex, and it may also occur in juvenile xanthogranuloma, sarcoidosis, leprosy, and other conditions.
Herpes simplex may incite a hypersensitivity reaction, which accounts for most of the tissue damage (85). In chronic or recurrent herpes simplex keratitis, viral cultures of corneal tissue are usually negative, and viral inclusions are rarely identified in tissue sections. Transmission electron microscopy, immunocytochemical methods, in situ hybridization, and the polymerase chain reaction (PCR) may be helpful in establishing the diagnosis in some cases (86).
RHEUMATOID ARTHRITIS
Individuals with rheumatoid arthritis are susceptible to a spontaneous thinning of the peripheral or central corneal stroma (87–89) associated with an ulcerative keratitis (90). Thinning of the peripheral cornea is more common, but the central corneal melts cause perforation more frequently. In some instances, a full-thickness perforation develops, whereas, in others, the stromal tissue loss occurs in the presence of an intact Descemet membrane (descemetocele).
EPITHELIAL INGROWTH
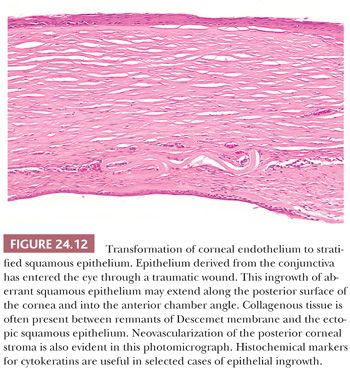
Sometimes after a penetrating corneal wound resulting from an accident, a cataract extraction, or another ocular surgical procedure, the epibulbar epithelium grows through the wound and into the anterior chamber (91,92). This may replace the corneal endothelium (Fig. 24.12), causing bullous keratopathy. It may also cause intractable glaucoma if the epithelium invades the trabecular meshwork. Epithelial ingrowth is easily diagnosed in pathologic specimens in which the normally single-layered corneal endothelium is replaced by several layers of squamous epithelium. In subtle cases, immunocytochemical staining for cytokeratin facilitates the diagnosis, because the endothelial cells lining the cornea and trabecular meshwork do not contain histochemically detectable keratin.
NEOPLASMS
Tumors of the cornea are rare, and they almost invariably represent the direct spread of squamous cell carcinoma or melanoma from the conjunctiva or eyelid.
SCLERA
From the standpoint of surgical pathology, most scleral specimens are related to inflammatory reactions (93). A necrotizing scleritis is often part of a systemic collagen autoimmune disease (94,95,96). Primary and metastatic tumors of the sclera are exceedingly rare (96), but uveal melanomas frequently spread by growing through the emissary channels in the sclera.
CONJUNCTIVA
Conjunctival tissue is usually biopsied for congenital abnormalities (97,98); inflammatory lesions, such as suspected sarcoidosis (99,100); tumors; or possible systemic metabolic diseases (101). Specimens from the conjunctiva are often extremely small, and communication between the surgeon and pathologist is therefore critical to ensure proper orientation when the precise orientation is important. The specimen should be spread out on a flat surface in the operating room, and the relevant landmarks should be labeled. When the surgical margins of resection must be evaluated, the specimen should be allowed to adhere to a level surface during fixation to prevent the specimen from curling.
DEVELOPMENTAL ABNORMALITIES
Nonneoplastic masses composed of tissues not normally found in conjunctiva sometimes form, especially at the junction of the conjunctiva and the cornea (corneoscleral limbus). Such choristomas include epibulbar dermoids, dermolipomas, and complex choristomas.
Epibulbar Dermoids
Epibulbar dermoids are characterized by dense, fibrocollagenous tissue containing sebaceous glands, hair follicles, and sweat glands, and they are covered by epidermis. They are frequently located in the inferotemporal conjunctiva. These choristomas are usually isolated lesions, but they are sometimes associated with other ocular abnormalities (colobomas of the iris and ciliary body), Goldenhar syndrome (pretragal auricular appendages, blind-ended preauricular fistulae, vertebral anomalies) (98), or the organoid nevus syndrome (linear nevus sebaceus of Jadassohn, Solomon syndrome) (102,103).
Dermolipomas and Complex Choristomas
Adipose connective tissue may constitute a major component of an epibulbar choristoma (dermolipoma). Less commonly, varying amounts of cartilage, lacrimal tissue, smooth muscle, adipose tissue, and even neural tissue are also present within the mass (complex choristomas).
CYSTS
Dermoid Cysts
Like dermoid cysts elsewhere, those of the conjunctiva are lined by a stratified squamous epithelium, and they contain cutaneous adnexal structures (104).
Inclusion Cysts
Inclusion cysts of the conjunctiva usually have a one-cell or two-cell lining of nonkeratinizing epithelium that contains goblet cells (105).
INFLAMMATION
Conjunctivitis is usually not a source of ophthalmic surgical pathology specimens. Certain changes in the conjunctival tissue in chronic conjunctivitis are nonspecific. The mucin-secreting goblet cells of the normal conjunctival epithelium often become numerous, and a hyperplastic epithelium may acquire papillary folds. Small islands of epithelium may become isolated and may form retention cysts that eventually calcify. In long-standing conjunctivitis, epithelial atrophy and keratinization with stromal scarring may occur.
Ligneous Conjunctivitis
The woody induration of the eyelid and conjunctiva that accompanies some cases of bilateral pseudomembranous conjunctivitis is occasionally excised, but it recurs relentlessly. The lesions in this rare ligneous conjunctivitis contain a considerable amount of fibrin but also immunoglobulins (106). The condition is caused by homozygous mutations in the PLG gene on human chromosome 6 (6q26) that encodes for plasminogen (107).
Ocular Cicatricial Pemphigoid
Ocular cicatricial pemphigoid, a mucocutaneous autoimmune disorder, frequently affects individuals older than 50 years of age. The condition affects women more often than men. The conjunctiva is involved in more than half of the cases, but this usually occurs 10 years after the onset of cutaneous or another mucosal disease (108,109). Epithelial erosions and bullae form in the conjunctiva early in the course of the disorder, but, later, the eye becomes scarred and dry.
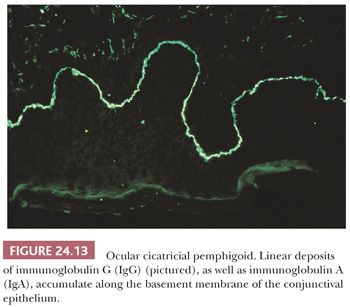
Conjunctival tissue from patients with suspected ocular cicatricial pemphigoid should be submitted in a fixative suitable for immunofluorescent studies. A pathognomonic linear deposition of immunoglobulin A (IgA) and IgG along the conjunctival basal lamina occurs in ocular cicatricial pemphigoid (Fig. 24.13). Light microscopy of conjunctival biopsies late in the course of the disease discloses nonspecific epithelial and stromal scarring, perivascular infiltrates of lymphocytes, plasma cells, and occasional eosinophils. A cicatricial conjunctivitis can be a paraneoplastic manifestation of a nonocular carcinoma (110).
Sarcoidosis
The ocular tissues are involved in up to 38% of patients with sarcoidosis (100). Because of its common involvement in sarcoidosis, a biopsy frequently is performed on the conjunctiva to establish a tissue diagnosis of sarcoidosis (99). When sarcoidosis is present, light microscopy reveals the typical nonnecrotizing granulomatous inflammation in the absence of stainable microorganisms. Because the granulomatous inflammation of sarcoidosis may be focal, step sections through the entire specimen are often indicated in cases of clinically suspected sarcoidosis.
Other Granulomatous Conjunctivitis
Other causes of granulomatous inflammation, such as tuberculosis, cat scratch fever (111,112), tularemia, syphilis, or other infections, and foreign bodies (113) may involve the conjunctiva, and these must be considered in the differential diagnosis. In contrast to sarcoidosis, the granulomatous inflammation in tuberculosis, cat scratch fever, and tularemia is characterized by extensive necrosis.
NEOPLASMS
Benign: Squamous Papilloma
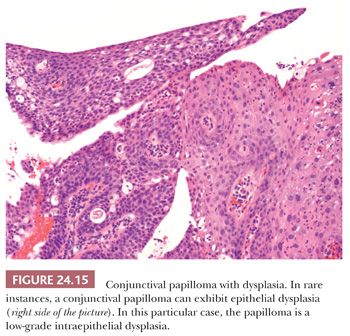
Squamous papillomas of the conjunctiva occur in diverse clinical settings, and they probably lack malignant potential (Fig. 24.14). In children, conjunctival papillomas are often bilateral, and they recur after excision (“recurrent conjunctival papillomatosis”). Characteristically, these pedunculated lesions are composed of papillomatous fronds of squamous epithelium that cover a fibrovascular core. Human papillomavirus has been implicated in the development of these lesions in younger individuals (114,115). It is reported that about 6% of the conjunctival papillomas present epithelial dysplasia (Fig. 24.15) (116).

In adults, papillomas are usually solitary and unilateral, and they may be confused clinically with squamous cell carcinoma. Inverted papillomas of the conjunctiva are rare (117).
Premalignant: Intraepithelial Neoplasms or Dysplasia and Intraepithelial Carcinoma
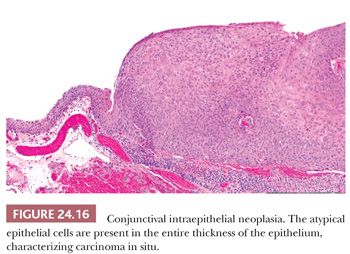
Dysplasia of the conjunctival epithelium is characterized by acanthosis, loss of cellular polarity, and cellular pleomorphism, and it resembles dysplasia of the uterine cervix microscopically. Depending on the extent of the epithelial abnormalities, conjunctival intraepithelial neoplasia can be designated as mild, moderate, or severe. In intraepithelial carcinoma, atypical cells extend throughout the entire epithelial thickness, but the lesion does not extend beneath the basal lamina of the conjunctival epithelium (Fig. 24.16). The adjacent corneal epithelium may become involved. Because dysplasia and intraepithelial carcinoma represent a spectrum of change, the nature of which depends on tissue sampling, these lesions are often designated ocular surface squamous neoplasia (118,119).
Malignant: Squamous Cell Carcinoma
Squamous cell carcinoma of the conjunctiva usually grows in a papillary or exophytic manner (Fig. 24.17). It is characterized histopathologically by cellular atypia throughout the entire thickness of the epithelium and by individual neoplastic cells or nests of tumor cells that extend into the underlying stroma. The epithelium is sometimes keratinized. When the carcinoma is large, it may invade the globe or the orbit (120); however, conjunctival carcinoma is rarely responsible for death.
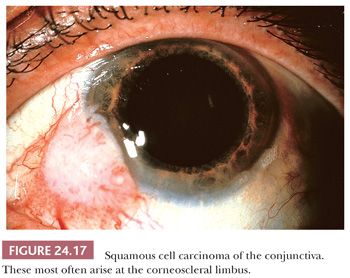
Occasionally, squamous cell carcinoma of the conjunctiva is jet black and, clinically, it mimics a melanoma. However, in contrast to malignant melanocytic neoplasms, the pigmented squamous cell carcinoma occurs in heavily pigmented individuals. Insufficient tumors of this type have been documented to evaluate their biologic behavior, but it seems to be the same as that of conjunctival nonpigmented squamous cell carcinomas (121).
Mucoepidermoid Carcinoma
Mucoepidermoid carcinoma of the conjunctiva is a rare but aggressive neoplasm that resembles squamous cell carcinoma in appearance but contains mucus-secreting cells and intraepithelial mucin. The mucin may not be readily apparent without the use of special stains, such as Alcian blue, Hale colloidal iron technique, or mucicarmine. This tumor must be differentiated from the more common squamous cell carcinoma of the conjunctiva, as it is more likely to invade the eye and orbit (122,123).
Spindle Cell Carcinoma
Spindle cell carcinoma rarely arises in the conjunctiva, but it pursues a more aggressive clinical course than the usual conjunctival squamous carcinoma (124). Like elsewhere, the tumor must be differentiated from spindle-shaped sarcomas, and both immunohistocytochemistry and transmission electron microscopy may be helpful in this regard. IHC staining of tissue sections discloses the presence of intracytoplasmic cytokeratin within the tumor, and ultrastructural studies reveal that tumor cells possess epithelial features, such as desmosomes and tonofibrils.
Melanocytic Lesions
Increased conjunctival melanotic pigmentation may be congenital or acquired, and, because various forms of conjunctival pigmentation are premalignant or malignant, they often create a diagnostic challenge for the pathologist (125,126). Acquired conjunctival melanosis may develop in previously normal eyes (primary acquired conjunctival melanosis), or it may be the result of inflammation or a neoplasm of the conjunctiva as well as of a metabolic (e.g., Addison disease) or a toxic state (secondary acquired conjunctival melanosis).
Nevocellular Nevi
Nevocellular nevi are common in the conjunctiva. They are frequently pigmented, but are not necessarily so, and they may involve the subepithelial tissues (subepithelial nevi), the subepithelium and epithelium (compound nevi), or the base of the epithelium (junctional nevi). In contrast to their counterparts in the skin, compound and subepithelial nevi are frequently associated with a substantial mononuclear inflammatory infiltrate in the conjunctival stroma and epithelial inclusion cysts. Occasionally, enlargement of these epithelial cysts may lead to the clinical suspicion of a conjunctival malignancy. The epithelial hyperplasia should not be confused with invasive squamous cell carcinoma. Junctional nevi are most commonly present in children.
Ephelis or Freckle
Congenital pigmentation of the conjunctival epithelium (ephelis or freckle) does not evolve into a melanoma.
Congenital Ocular Melanocytosis and Oculodermal Melanocytosis (Nevus of Ota)
Congenital discoloration of the subepithelial tissues of the conjunctiva may be associated with congenital pigmentation of the uvea and other parts of the eye (ocular melanocytosis), and a benign clinical course usually ensues. If the skin of the eyelids or the periorbital area is also affected, the condition is known as oculodermal melanocytosis (nevus of Ota), which may carry a very slight risk of uveal melanoma, estimated at 1 in 400 affected patients. However, patients who develop melanoma were found to have double the risk for metastasis compared to those without melanocytosis (127).
PREMALIGNANT: PRIMARY ACQUIRED MELANOSIS
Primary acquired melanosis (PAM) is a unilateral acquired variety of conjunctival pigmentation that slowly affects the conjunctiva in middle-aged people of European ancestry. It accounts for 11% of the conjunctival tumors (128). PAM is characterized by an evolving spectrum of varying degrees of intraepithelial melanocytic hyperplasia (benign acquired melanosis) or dysplasia (melanocytic dysplasia) and a variable subepithelial mononuclear cell infiltrate and vascular engorgement. Biopsies are occasionally performed on the lesions to establish a tissue diagnosis or because of the clinical suspicion of a melanoma. Tissue sampling is important in evaluating the conjunctiva because different parts of the same conjunctiva may manifest different degrees of the disorder, and parts may portray melanoma (129). PAM with and without atypia is characterized by the presence or absence of atypical melanocytes, respectively.
When the individual atypical melanocytes are confined to the basal epithelium, the patient’s risk of developing melanoma is about 20% (129). However, if nests or pagetoid spread of atypical cells is present, approximately 90% of such lesions eventually develop into a melanoma (129). In a different study, recurrence was found in 11% of the PAM without atypia, 26% with mild atypia, and 50% with severe atypia. Progression to melanoma occurred in 13% of the PAM with severe atypia and none of the PAM with no cytologic atypia or mild atypia (128).
MALIGNANT
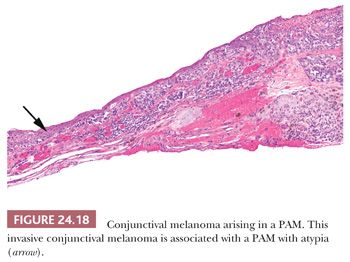
Conjunctival melanomas are uncommon, and they are typically pigmented. They may arise from PAM, nevocellular nevi, or apparently normal conjunctiva, but the nature of the initial lesion does not seem to be of prognostic importance. It has been estimated that approximately 75% of conjunctival melanomas arise in association with PAM (128) (Fig. 24.18). Conjunctival melanomas are often apparently multicentric, especially when they are preceded by PAM. In a series of 131 patients with conjunctival melanomas followed up for a median period of at least 8 years, about 25% of patients died from metastases (130).
LYMPHOID TUMORS
Reactive Lymphoid Hyperplasia
Reactive lymphoid hyperplasia may be difficult to distinguish from lymphoma. Features suggestive of reactive lymphoid lesions include a population of well-differentiated lymphocytes with occasional plasma cells, macrophages, eosinophils, and germinal follicles, which are often irregular in shape and distribution, within a stroma that contains scant fibrous tissue. These reactive germinal follicles often contain tingible bodies in macrophages and significant mitotic activity.
Although some lymphocytes are present in the conjunctiva of apparently healthy persons, a large number of lymphocytes (especially when they are arranged in lymphoid follicles with germinal centers) suggests reactive lymphoid hyperplasia. Other features of reactive conjunctival lymphoid hyperplasia include a prominent vascularity with hyperplastic, plump vascular endothelial cells and a polymorphic, immunophenotypically polyclonal population of mononuclear cells (131).
Lymphoma
Lymphocytic proliferations in the conjunctiva typically produce salmon-colored masses; from the standpoint of therapy and prognosis, these lesions remain an enigma to the clinician and pathologist.
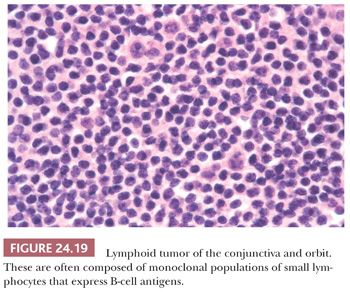
The lymphoid tissue of the conjunctiva and, arguably, also of the orbit forms part of the mucosa-associated lymphoid tissue (MALT). The conjunctiva-associated lymphoid system consists of immunocompetent cells together with specialized conjunctival epithelial and dendritic antigen-presenting cells. Most lymphomas arising from the conjunctiva and orbit resemble other MALT-derived lymphomas. Therefore, the most common type of lymphoma affecting these areas is the marginal zone B-cell lymphoma (132). They remain localized for a long time, and they are often preceded by an apparent reactive inflammatory stage. Conjunctival and orbital lymphomas are usually composed of well-differentiated small lymphocytes, and they occur as isolated lesions or in association with a systemic lymphoma. They are usually composed of a monoclonal population of B lymphocytes (Fig. 24.19) (133), and fewer than 15% are of the follicular or nodular type. Less commonly, diffuse large B-cell lymphoma and mantle cell lymphoma can arise in these areas (132). The cell population of orbital lymphomas is rarely derived from T cells (134). Orbital lymphomas usually express a restriction of κ-light or λ-light chains; although monoclonal lymphomatous masses are generally regarded as neoplastic, not all of those in the orbit or conjunctiva progress to systemic disease. Furthermore, some polyclonal masses that are considered to represent benign reactive lymphoid hyperplasia by an immunocytochemical evaluation express B-lymphocyte clonality on molecular genetic analysis (135). Orbital involvement in Burkitt lymphoma (136) is common in Africa, but the orbit is rarely involved in Hodgkin disease (137).
These conjunctival lymphoid tumors are difficult to compartmentalize as benign or malignant. Their malignant potential is often difficult, if not impossible, to predict by using routine light microscopy, IHC (138), and even molecular biologic studies for immunoglobulin and T-cell receptor gene rearrangements (139). Most lymphomas of the conjunctiva are first seen as isolated lesions, and remain so; sometimes, however, an associated systemic lymphoma exists at the time of the conjunctival presentation or on subsequent follow-up. Several genetic abnormalities have been described in MALT lymphomas (140). Like elsewhere, when a lymphoma of the orbit or conjunctiva is suspected because of the clinical appearance or as a result of a histopathologic evaluation with frozen sections at the time of surgery, fresh tissue from conjunctival and orbital lymphoid tumors can be appropriately prepared for the immunohistopathologic evaluation of the lymphocyte populations, flow cytometry, and molecular biologic analysis of immunoglobulin and possibly B- and T-cell receptor gene rearrangement.
OTHER TUMORS
Benign
Other benign tumors of the conjunctiva include hereditary benign intraepithelial dyskeratosis (141).
Malignant
Other malignant neoplasms that occasionally arise in the conjunctiva include rhabdomyosarcoma (142); leiomyosarcoma (143); and Kaposi sarcoma, which most frequently develops in patients with AIDS (144). Malignant neoplasms of the orbit and eyelid may also extend to the conjunctiva.
Nonneoplastic Growths
Pinguecula. Extensive conjunctival elastosis can produce an elevated whitish-yellow lesion near the corneal border. Because of its clinical resemblance to fat, this mass is called a pinguecula. It is rarely removed surgically; when it is, it is mainly for cosmesis. A prominent foreign body giant cell reaction sometimes surrounds and incorporates the elastotic material (145). Such an actinic granuloma may lead the pathologist to suspect microorganisms.
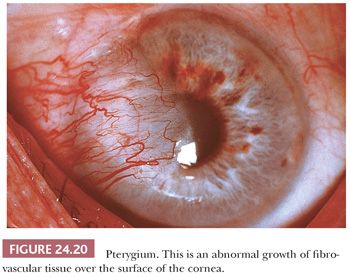
Pterygium. Vascularized conjunctival tissue may grow horizontally over the cornea in a triangular fashion. In most parts of the world, this pterygium is almost always associated with an underlying actinic elastosis of the conjunctiva (146), and it is thought to be the result of long-term solar (actinic) irradiation (Fig. 24.20).
Pterygia are often excised when they compromise the visual axis or induce corneal astigmatism, and occasionally when they are unsightly. Pterygia frequently recur after excision but this is not indicative of malignant transformation. Histologically, pterygia are composed of vascularized connective tissue, usually with solar elastosis and a peculiar corkscrew configuration of individual collagen fibers. In the cornea, the Bowman layer may be focally destroyed. The contiguous corneal epithelium may be atrophic or thickened and dysplastic. The conjunctival epithelium frequently contains more mucus-secreting goblet cells than is normal, and, in keeping with its history of being sun-induced, it is occasionally dysplastic or it may have intraepithelial carcinoma. Cases of conjunctival intraepithelial neoplasia (147) and squamous cell carcinoma mimicking or arising in pterygia have been reported (148).
CARUNCLE
The caruncle is the fleshy nodular prominence in the nasal portion of the interpalpebral fissure. It is lined by conjunctival epithelium, and it contains cutaneous adnexal structures. Sebaceous gland hyperplasia (149), sebaceous carcinoma (150), oncocytoma (151), basal cell carcinoma (152), and other tumors arise from the caruncle in rare instances. Conjunctival tumors may extend into the caruncle.
EYELIDS
Several pathologic processes involving the eyelids are treated by the ophthalmologist, and a high percentage of surgically excised ophthalmic specimens submitted for histopathologic evaluation are obtained from this site. Tissue excised during cosmetic blepharoplasty, blepharoptosis repair, or other reparative eyelid surgery may also be submitted for pathologic evaluation, but this does not usually disclose any noteworthy abnormalities. Surgically excised tissue from the eyelid in the floppy-eyelid syndrome (153) does not have any specific light-microscopic features. Other cutaneous disorders affecting the eyelid are identical to those found elsewhere in the skin; these are discussed in Chapters 1, 2, and 3. As in other parts of the body, certain tumors of the eyelid, such as keratoacanthoma and sebaceous tumors, may be associated with visceral carcinomas, especially of the colon (Muir-Torre syndrome) (154,155).
INFLAMMATORY REACTIONS
Chalazion
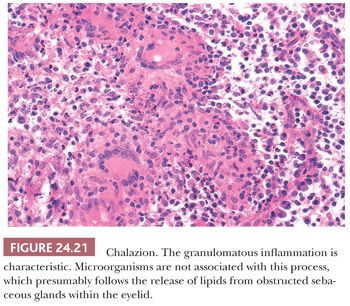
A chalazion is a localized, acute, lipogranulomatous, inflammatory process within the eyelid that is sometimes encapsulated. After an obstruction of any cause, the ducts draining the sebaceous glands (meibomian glands) within the eyelid may rupture, releasing material that presumably incites the reaction. Several factors, including inspissated secretions, infections, or neoplasms, may lead to the ductal obstruction. Specimens submitted for pathologic examination are usually curettings and sometimes biopsies of discrete masses. Most ophthalmologists do not submit suspected chalazia for pathologic evaluation after initial excision or curettage; however, because chalazia and sebaceous carcinoma are sometimes indistinguishable clinically, a histopathologic evaluation is imperative for recurrent chalazia (156). The characteristic histopathologic feature is a mixed inflammatory cell infiltrate composed of polymorphonuclear leukocytes, lymphocytes, plasma cells, macrophages, epithelioid cells, and, often, multinucleated giant cells (Fig. 24.21). Although this microscopic appearance may resemble that of other granulomatous processes, such as sarcoidosis, tuberculosis, or fungal infections, special stains for microorganisms are usually not justified in the typical case in view of the low yield of specific causal agents for the cost.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


