CHAPTER 16
Extended-Spectrum Triazole Antifungals: Posaconazole and Voriconazole
KELLY E. MARTIN, PharmD, BCPS
MAURICE ALEXANDER, PharmD, BCOP, CPP
BENYAM MULUNEH, PharmD, BCOP, CPP
OVERVIEW
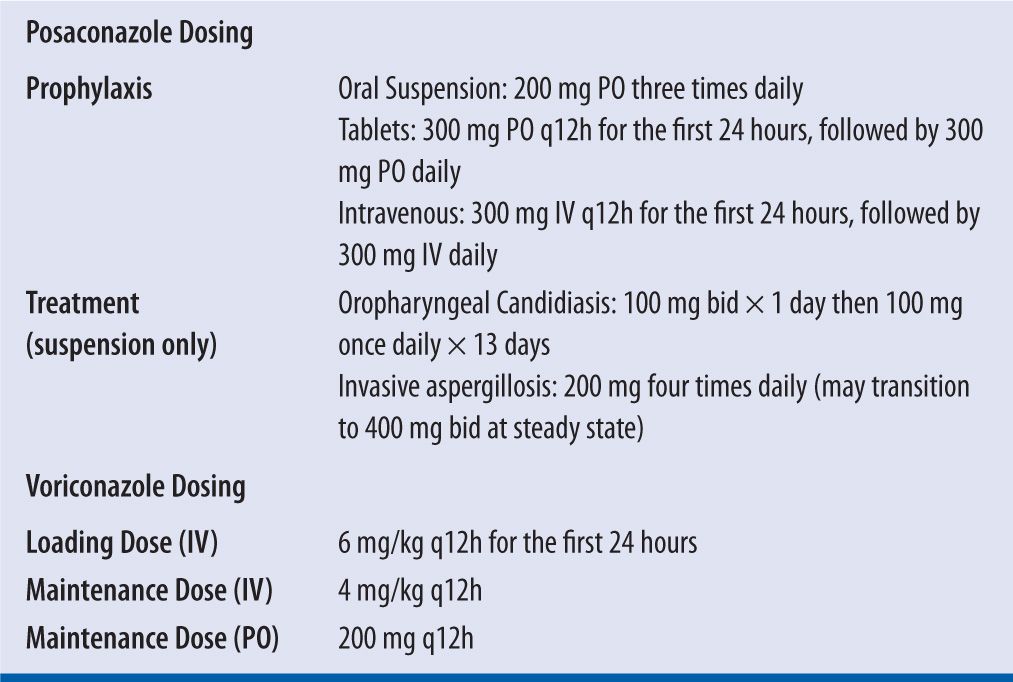
Voriconazole and posaconazole are broad-spectrum triazole antifungal agents. They decrease ergosterol synthesis by interfering with the lanosterol-14α-demethylase (P450 enzyme) activity leading to a malformation of the fungal cell membrane. These agents have activity against most yeasts, such as fluconazole-resistant Candida spp. and molds, such as Aspergillus spp. and Fusarium spp. Unlike voriconazole, posaconazole has activity against the Mucorales order.1,2 Posaconazole and voriconazole play a significant role in both the prevention and treatment of opportunistic invasive fungal infections, especially in immunocompromised patients. Because of its wide spectrum of activity, posaconazole is indicated for prophylaxis of invasive Aspergillus and Candida infections in patients who are at high risk of developing these infections due to prolonged immunosuppression after stem cell transplant or prolonged neutropenia after chemotherapy for a hematologic malignancy.1,3 Voriconazole is considered the drug of choice for treatment of most invasive aspergillosis infections.2,4 Voriconazole is also approved for use in nonneutropenic candidemia and as salvage therapy for Scedosporium apiospermum and Fusarium spp. infections.2 Although posaconazole is FDA-indicated for the treatment of refractory oropharyngeal candididasis, the suspension formulation also has positive data to support its use at higher doses for the treatment of other invasive fungal infections including mucormycosis and cryptococcal infections.5,6 Refer to Table 16-1 for FDA-approved treatment and prophylactic dosing recommendations for voriconazole and posaconazole. To date, the data for posaconazole delayed-release (DR) tablets and intravenous formulations are limited to prophylactic indications.1 Voriconazole and posaconazole therapeutic drug monitoring (TDM) can be utilized to improve patient outcomes and, in the case of voriconazole, to limit toxicity.
| TABLE 16-1 | Pharmacokinetic Parameters of Voriconazole and Posaconazole1,2,8,12,15,31,46,47 |
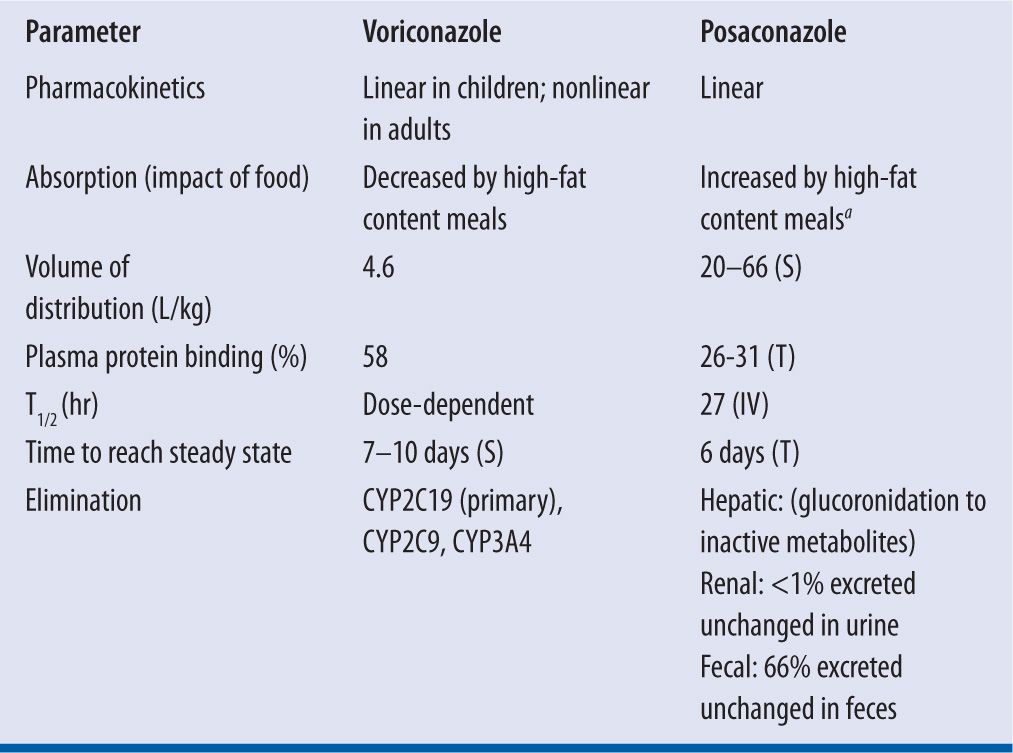
aPosaconazole suspension, 4-fold increase in Cmax and AUC; Posaconazole tablets, 16% increase in Cmax and 51% increase in AUC
S, suspension; T, tablets: IV, intravenous
BIOAVAILABILITY (F)
Posaconazole oral suspension has variable bioavailability that is significantly influenced by dose and food intake. This formulation has saturable absorption requiring a smaller, multiple daily dosing schedule despite the drug’s long half-life. For example, dosing posaconazole 400 mg every 12 hours versus 800 mg once doubles the bioavailability and dosing it 200 mg every 6 hours increases it by nearly threefold. These evaluations, however, were done in patients in the fasted state.7 Administration of posaconazole oral suspension with a high-fat meal has been shown to increase the bioavailability fourfold leading to the current recommendation to take posaconazole oral suspension with a fatty meal.8 Of note, the absorption of posaconazole oral suspension is also influenced by suppression of gastric acid secretion by proton-pump inhibitors and prokinetic agents such as metoclopramide.1 In 2013, the FDA approved a delayed-release tablet formulation of posaconazole. While there is less variability compared to the suspension, the tablets should also be taken with food. Pharmacokinetic studies have shown that a high-fat meal increases the AUC by 51%. Unlike the suspension, the tablet formulation absorption is not affected by gastric acid suppressors or gastrointestinal motility agents.1,9 Of note, an intravenous formulation of posaconazole was approved by the FDA in 2014.
Voriconazole is also available in both oral and intravenous formulations. In healthy subjects, voriconazole has 96 percent oral bioavailability with the time to maximum plasma concentration (Cmax) ranging from 1–2 hours after administration.2 Oral voriconazole should be administered in the fasting state.2,10 Ingestion of voriconazole following a high-fat meal resulted in a mean decrease in Cmax of 34 percent and 58 percent with the tablet and oral suspension, respectively.2 Unlike posaconazole, medications that increase gastric pH have not been found to impact voriconazole absorption.
VOLUME OF DISTRIBUTION (V)
Both voriconazole and posaconazole have a large volume of distribution, with extensive distribution from the plasma into tissues. After absorption, posaconazole has a volume of distribution that ranges from 5 L/kg to 25 L/kg, allowing substantial tissue penetration.8 Cerebrospinal fluid (CSF) drug levels are low compared to serum levels (CSF to plasma ratio of 0.004–0.009), suggesting poor penetration into the central nervous system (CNS). However, it is postulated that patients with CNS fungal infections have a potentially compromised blood-brain barrier leading to increased posaconazole penetration and positive clinical outcomes.11
At steady state, voriconazole has a volume of distribution estimated to range from 2 L/kg to 4.6 L/kg, suggesting extensive tissue distribution.2,12 Voriconazole is able to penetrate the blood-brain barrier and achieve concentrations in the CSF of approximately 50 percent of the plasma concentrations.12 However, voriconazole concentrations in the brain may be even higher than the CSF as shown in an autopsy study of eight patients.13 Animal studies have shown voriconazole concentrations to be significant in the pulmonary epithelial lining fluid, which may be of importance for the treatment of pulmonary fungal infections. Voriconazole also distributes into intracellular components including the polymorphonuclear leukocytes (PMNs). It has been demonstrated that concentrations within the PMNs may be up to 8.5 times greater than the concentrations found in the plasma.12
CLEARANCE (CL)
Posaconazole and voriconazole differ substantially in how they are cleared from the body. About 15 percent of posaconazole is metabolized through glucoronidation, avoiding the cytochrome P450 (CYP) pathway. Although posaconazole is not metabolized by the P450 (CYP) pathway it is a potent inhibitor of CYP3A4.11,14 P-glycoprotein has also been implicated in the excretion of posaconazole because of the increased drug concentrations observed with inhibition of this enzyme.8 Fecal excretion of the parent drug accounts for 77 percent of the drug’s elimination, with renal excretion playing a minor role (metabolites: 13–14%; parent: negligible).
Voriconazole is both a substrate and inhibitor of the CYP enzyme system. It is primarily metabolized by CYP2C19, and to a lesser extent metabolized by CYP3A4 and CYP2C9.2 Voriconazole displays nonlinear pharmacokinetics likely due to saturable metabolism, meaning that an increase in dose results in a disproportionate increase in plasma concentration.12,15 The major metabolite of voriconazole, which accounts for almost three-quarters of all voriconazole metabolites, is N-oxide. N-oxide has minimal antifungal activity and an unknown impact on toxicity.2,12,16
The CYP2C19 isoenzyme exhibits genetic polymorphism, which results in poor and extensive metabolizer phenotypes and may account for almost 50 percent of the variability in voriconazole clearance2,12,16,17 The poor metabolizer phenotype is found in 15–20 percent of Asian populations and 3–5 percent of Caucasian and Black populations. Those with the poor metabolizer phenotype can have up to fourfold higher voriconazole concentrations compared to the homozygous extensive metabolizer phenotype.2,16 Despite this known variability, no current specific dosing recommendations are based on CYP2C19 genotype.2
Less than 2 percent of voriconazole is excreted in the urine and no dose adjustments are required in renal dysfunction. However, in both posaconazole and voriconazole IV formulations, the vehicle, sulfobutyl ether beta-cyclodextrin sodium (SBECD), can accumulate in patients with creatinine clearance (CrCl) <50 mL/min.1,2,18
ELIMINATION HALF-LIFE (T1/2)
Due to the nonlinear pharmacokinetics, voriconazole has dose-dependent elimination. The mean elimination is estimated to range 6–9 hours and increases after multiple doses compared to single dose administration.2,12,15
Posaconazole also had a dose-dependent half-life, ranging 20-66 hours, which is decreased by prior hematopoietic stem cell transplantation and varies based on formulation administered.1,7,11,19 Table 16-2 summarizes the pharmacokinetic parameters of voriconazole and posaconazole.
| TABLE 16-2 | Voriconazole and Posaconazole Dosing1,2 |
THERAPEUTIC CONCENTRATIONS
A voriconazole exposure-response relationship has been established through studies in which lower serum concentrations were associated with a lack of clinical response, progression of fungal infection, or increased breakthrough fungal infections. Although no consensus sets an exact lower limit of the therapeutic range, studies have targeted voriconazole concentrations based on the minimum inhibitory concentrations (MIC) of clinically relevant fungal pathogens. Most Candida and Aspergillus species have an MIC90 of 0.25–1 mg/L.20,21 In general, treatment success has been associated with voriconazole trough concentrations >1 mg/L, although some studies suggest targeting troughs ≥2 mg/L for maximal efficacy.22-24
Because of the erratic absorption and variable half-life, determining a serum concentration-to-effect relationship is important for posaconazole. In patients who are critically ill or have undergone stem cell transplantation, absorption can be decreased, leading to variability in serum concentrations secondary to altered gastric mucosa and/or decreased appetite.15 A number of studies have attempted to characterize the concentration-efficacy relationship. The authors of one study that evaluated posaconazole in the treatment of invasive aspergillosis found 75 percent of patients responded with an average serum concentration (Cavg) of 1,250 ng/mL compared with only 24 percent that responded with a Cavg of 134 ng/mL.25 Based on these findings, it is recommended to target a concentration greater than 1,000–1,250 ng/mL when treating invasive fungal infections.26,27
Studies looking at the prophylactic effects of posaconazole in immunocompromised patients have not found a similar concentration-effect relationship.3,28 However, a clinical pharmacology review and logistic regression by the Food and Drug Administration (FDA) found achieving a Cavg of less than 700 ng/mL was strongly predictive of an invasive fungal infection.29
TOXIC CONCENTRATIONS
Voriconazole trough concentrations should be maintained below an upper limit of 5–5.5 mg/L to minimize adverse events including hepatotoxicity and neurotoxicity.22-24
A posaconazole exposure-toxicity relationship remains to be established. A logistic regression of two separate trials by the FDA found that patients with lower serum concentrations (Cavg = 205 ± 105 ng/mL) developed fewer toxicities compared to patients with higher concentrations (Cavg = 1,751 ng/mL ± 538 ng/mL). However, no statistical difference or definitive conclusion was made regarding a concentration beyond which posaconazole would be considered toxic.29
DRUG LEVEL MONITORING
Monitoring of voriconazole concentrations is recommended for assessment of both efficacy and toxicity. Variability in voriconazole concentrations is multifactorial due to nonlinear pharmacokinetics, CYP2C19 genotype polymorphisms, underlying hepatic dysfunction, drug-drug interactions as well as still undefined factors. For example, a recent study suggested that oral voriconazole doses are not equivalent to IV voriconazole doses despite the high bioavailability initially described in the package insert.30 In clinical practice, most patients should receive voriconazole therapeutic drug monitoring.
Because of the interpatient variability in absorption in most patients—including those with graft-versus-host disease, diarrhea, compromised mucosal barriers, decreased oral intake limiting administration with food, and gastric acid suppression—checking plasma concentrations of posaconazole is recommended to assess for efficacy. This is particularly true of the posaconazole suspension for which absorption is most variable. It is theorized that serum concentrations may not always be accurate in predicting the therapeutic efficacy of posaconazole because of the high volume of distribution leading to a higher concentration in tissues rather than in plasma.15 Although not incorporated in clinical practice, concentrations in isolated alveolar cells and intracellular measurements of neutrophils and monocytes are 22–67 times larger than plasma concentrations.26
TIME TO DRAW LEVELS AND FREQUENCY, MONITORING
Voriconazole trough concentrations are used clinically to monitor for efficacy and toxicity. Due to the nonlinear kinetics, it is important that voriconazole trough concentrations are measured at the end of the 12-hour dosing interval (30 minutes to 1 hour prior to the next dose). No mathematical equations are available to extrapolate a random serum concentration that is drawn during the middle of the dosing interval. Concentrations should be measured once steady state is reached 5–7 days after the maintenance dose has been initiated.15 Although steady-state concentrations can be reached in 1–2 days following a loading dose, it is generally considered prudent to wait at least 5 days to measure a trough concentration because of variability in the elimination half-life that also requires time to reach steady-state. As mentioned earlier, voriconazole has saturable metabolism and the mean elimination half-life increases after multiple doses.12,15 It is also recommended to obtain a voriconazole trough concentration at any time during therapy if clinical failure is suspected or signs of toxicity are exhibited.
Due to posaconazole’s long half-life, the frequent dosing of the oral suspension, and the delayed-release properties of the tablet formulation, serum concentrations can be drawn at any time after the drug has reached steady state (7–10 days). Some limited evidence suggests that measuring levels earlier (at 3 days) may be predictive of steady-state concentrations.26 Because it takes about one week to reach steady state and several more days for most institutions to report level results, it may be beneficial to assess a level earlier to prevent delay in responding to a level that is subtherapeutic. However, levels should be reassessed once steady state is reached.
CASES
CASE 1: VORICONAZOLE INTRAVENOUS (IV) LOADING DOSE AND MAINTENANCE DOSE
JS is a 53-year-old African American male with a history of HIV (most recent CD4 count <20 cells/mm3) who was admitted for right-sided flank pain. He was found to have a renal abscess that was drained and sent for culture. The microbiology lab reported Aspergillus fumigatus. The patient is not currently compliant with their antiretroviral (ARV) regimen. Calculate a voriconazole IV loading dose followed by a maintenance dose.
Height = 6′
Weight = 185 lbs
Step 1: Calculate IV loading dose.
The package insert recommends administration of an IV loading dose of 6 mg/kg for 2 doses given 12 hours apart.
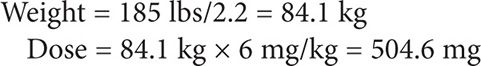
For ease of IV preparation, the dose can be rounded to 500 mg every 12 hours for the first 24 hours.
Step 2: Calculate the IV maintenance dose.
After 24 hours, the dose should be reduced to the maintenance dose of 4 mg/kg every 12 hours administered intravenously.
Dose = 84.1 kg × 4 mg/kg = 336.4 mg
The dose can be rounded to voriconazole 300 mg IV every 12 hours with the recommendation to check a trough concentration on day 5-7.
CASE 2: VORICONAZOLE IV TO ORAL (PO) CONVERSION
SM is a 48-year-old Caucasian female with chronic graft-versus-host disease on prolonged prednisone treatment who is admitted to the hospital for a productive cough for the past two weeks. Chest CT and galactomannan antigen testing suggest possible invasive pulmonary aspergillosis.
Height = 5′1″
Weight = 83 lbs
She is initially given a loading dose of IV voriconazole 6 mg/kg on day 1 and then started on voriconazole 150 mg IV every 12 hours. Determine an oral maintenance dose of voriconazole for this patient.
Step 1: Calculate weight in kilograms
Weight = 83 lbs/2.2 = 37.7 kg
Weight is important for oral dosing despite the fact that the FDA approved dosing is only weight-based when dosing intravenous voriconazole. The package insert recommends that adult patients who weigh less than 40 kg should receive half of the oral maintenance dose.
Step 2: Determine an oral maintenance dosing regimen for treatment of invasive aspergillosis infection
Two different methods may be used to determine a voriconazole regimen for the treatment of invasive aspergillosis using the oral formulation. The regimen can be determined using the FDA-approved fixed dosing or using weight based dosing (similar to the dosing for the IV formulation).
Although the FDA-approved recommendation is to utilize standardized doses of 200 mg every 12 hours when administering oral voriconazole, data show that this dose provides similar exposure to a 3 mg/kg IV dose. To achieve similar exposure to a 4 mg/kg IV dose, the equivalent oral dose is 300 mg.2,32 One concern with using the 200 mg oral dose is the possibility that this dose will not achieve steady-state concentrations greater than the MIC of Aspergillus species in most patients. In a pharmacokinetic study of 42 healthy male subjects, the 200 mg oral dose did not maintain troughs greater than 1 mg/L and thus fell within subtherapeutic concentrations.32 Additionally, a study using pharmacokinetic modeling demonstrated that oral voriconazole doses result in lower trough concentrations when compared to equivalent intravenous doses.30 For example, the likelihood of achieving a voriconazole trough concentration >1 mg/L with a 200 mg intravenous dose and 200 mg oral dose was 86 percent and 60 percent, respectively.30 Interestingly, this study suggests a lower oral bioavailability of about 60 percent than what has been reported in the package insert. Because this patient is being treated for an invasive fungal infection, it is necessary to achieve adequate trough concentrations for maximum efficacy. Based on the literature, concentrations are most likely achieved with an oral voriconazole dose of 300 mg every 12 hours.2,30,32 However, this patient is less than 40 kg and thus the dose should be reduced by 50 percent to 150 mg PO every 12 hours.2
Alternatively, many practitioners utilize oral weight-based dosing for treatment of invasive fungal infections. For this patient who weighs 37.7 kg, a 4 mg/kg oral dose would also result in dosing voriconazole at 150 mg PO every 12 hours. After 5–7 days of oral dosing, a trough voriconazole plasma concentration should be measured.
CASE 3: SUPRATHERAPEUTIC VORICONAZOLE TROUGH CONCENTRATIONS
DP is a 31-year-old Asian male who was admitted for an autologous hematopoietic stem cell transplant (HSCT). On day 3 following HSCT, he developed febrile neutropenia and was started on broad-spectrum empiric antibiotics with cefepime and vancomycin. After 96 hours, DP remained febrile at which time empiric antifungal treatment was initiated. He was given a loading dose of voriconazole 6 mg/kg IV every 12 hours for two doses and then started on voriconazole 200 mg PO every 12 hours. On day 6 of voriconazole therapy, a trough plasma concentration was drawn 30 minutes before the next dose was to be given. The patient denied any visual changes or disturbances. Determine how to interpret and respond to the voriconazole plasma trough concentration.
Height = 5′10″
Weight = 77 kg
Pertinent medications: omeprazole 40 mg PO daily
Voriconazole trough: 7.2 mg/L (therapeutic range: 1.0–5.5 mg/L)
AST: 20 IU/L
ALT: 15 IU/L
Total bilirubin: 0.8 mg/dL
Step 1: Identify possible factors that may have resulted in a supratherapeutic trough concentration.
Voriconazole has significant inter- and intrapatient variability, and several factors may contribute to trough concentrations that fall outside of the therapeutic range. It is important to consider CYP2C19 genotype polymorphisms, drug-drug interactions, timing of the trough concentration, and liver function.
• CYP2C19 Genotype: In clinical practice, the CYP2C19 genotype is often unknown and not a test that is routinely ordered prior to initiation of voriconazole therapy. However, this patient is known to be of Asian descent, which means a 15–20 percent likelihood he carries the CYP2C19 polymorphism that results in the “poor metabolizer” phenotype. As discussed earlier, this phenotype could result in voriconazole concentrations fourfold greater than the “wild type” phenotype.
• Drug-Drug Interactions: Medications that are CYP2C19 inhibitors, such as oral contraceptives containing ethinyl estradiol, can result in increased voriconazole plasma concentrations. In this case, the patient is receiving omeprazole 40 mg PO daily, which is a known CYP2C19 inhibitor. Studies conflict regarding the clinical impact of this drug-drug interaction.2,22,30,33-35 With the supratherapeutic trough, it would be prudent to change therapy from omeprazole to an H2-receptor antagonist, if the omeprazole is not specifically indicated. Of note, the package insert does not recommend any initial adjustments to voriconazole dose when coadministered with omeprazole.2 Therefore, omeprazole does not need to be discontinued when voriconazole is coadministered, unless it is suspected the drug-drug interaction is leading to supratherapeutic voriconazole concentrations or voriconazole toxicities.
• Liver function: In this case, the patient has normal liver function tests (LFTs), making it unlikely to be a factor contributing to the supratherapeutic voriconazole concentration. As voriconazole is hepatically metabolized, diminished liver function can result in reduced clearance of the drug.
Step 2: Determine an appropriate voriconazole dose adjustment based on the plasma trough concentration.
Voriconazole dose adjustments based on therapeutic drug monitoring have not been well-defined. With a supratherapeutic concentration, the first thing to consider is whether the patient is experiencing any toxicity. In this case, the patient does not have visual disturbances and LFTs are within normal limits. Due to the nonlinear pharmacokinetics and other potential factors that may be affecting the plasma concentrations such as CYP2C19 polymorphisms, it is difficult to predict the impact of a dose adjustment on the resulting concentration. The package insert recommends reducing the dose by 50 mg increments.2 A decrease to oral voriconazole 150 mg every 12 hours followed by another trough concentration in 5–7 days would be appropriate. Studies have shown significant intrapatient variability, so some practitioners, in the absence of toxicities, would consider repeating the trough concentration or waiting 5–7 days following discontinuation of the omeprazole and then repeating the trough concentration.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


