Ewing Sarcoma/Primitive Neuroectodermal Tumor (Pnet)
G. Petur Nielsen, MD
Andrew E. Rosenberg, MD
Key Facts
Terminology
Malignant small round cell sarcoma showing variable degree of neuroectodermal differentiation
Clinical Issues
Accounts for approximately 6-10% of primary malignant bone tumors
Usually arises in diaphysis or metadiaphysis of long tubular bones and flat bones of pelvis
Usually treated with combination of chemotherapy and surgery
Effective chemotherapy has improved prognosis from 5-15% to 75% 5-year survival
Image Findings
Destructive and lytic
Macroscopic Features
Tan-white, fish flesh-like in appearance; usually transgresses cortex and periosteum, producing soft tissue mass
Microscopic Pathology
Tumor cells are 1-2x size of lymphocytes
Tumor cells grow in sheets or irregular islands delineated by dense fibrous tissue
Architectural features suggestive of neural differentiation include organoid growth pattern and Homer Wright rosettes
Approximately 85% of neoplasms harbor t(11;22) (q24;q12) translocation
Cells expresses vimentin, CD99 (90%), and FLI-1 (90%)
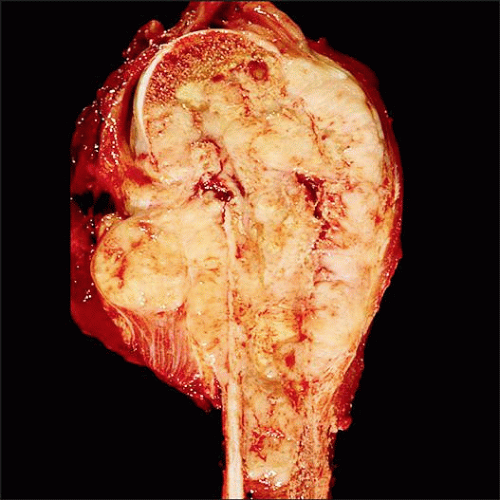 Ewing sarcoma is shown arising in the metadiaphysis of the humerus manifests as a fleshy mass centered in the medullary cavity, breaking through the cortex and forming a circumferential soft tissue mass. |
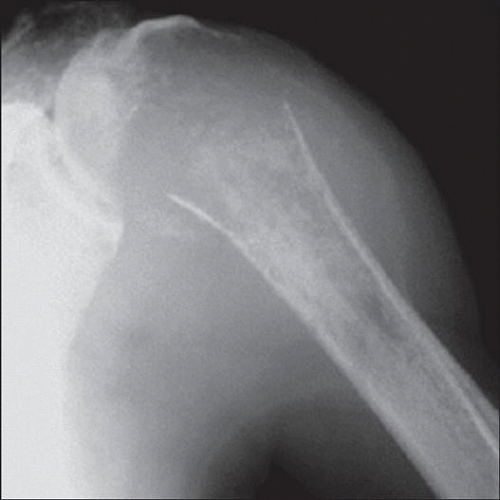 Radiograph of a Ewing sarcoma shows a highly aggressive and ill-defined destructive mass in the proximal humerus. The mass has broken through the cortex and extended into the adjacent soft tissues. |
TERMINOLOGY
Abbreviations
Ewing sarcoma (EWS)
Primitive neuroectodermal tumor (PNET)
Synonyms
Peripheral neuroepithelioma
Askin tumor
Definitions
Round cell sarcoma showing variable degree of neuroectodermal differentiation
Undifferentiated tumors have been diagnosed as Ewing sarcoma
Tumors demonstrating neuroectodermal differentiation by light microscopy, immunohistochemistry, or electron microscopy have traditionally been labeled PNET
ETIOLOGY/PATHOGENESIS
Neoplasm
Evidence suggests stem cell origin
CLINICAL ISSUES
Epidemiology
Incidence
Accounts for 6-10% of primary malignant bone tumors
Follows osteosarcoma and chondrosarcoma in frequency in adults, and osteosarcoma in children and adolescents
Age
Most patients between 10 and 15 years of age
Approximately 80% are younger than 20 years
Gender
Boys affected more frequently than girls (1.3-1.4:1)
Ethnicity
Striking predilection for whites (3/1,000,000); Africans, Asians, and Native Americans rarely affected (0.2/1,000,000)
Site
Can arise in any portion of skeleton
Usually develops in diaphysis or metadiaphysis of long tubular bones and flat bones of pelvis
Approximately 22% of cases arise in femur followed by ilium, tibia, humerus, fibula, and ribs
Tumors that originate in chest wall have been called Askin tumor
Presentation
Painful enlarging mass
Affected site is frequently tender, warm, and swollen
Some patients have systemic findings that mimic infection: Fever, elevated sedimentation rate, anemia, and leukocytosis
Pathologic fracture is an uncommon complication
Treatment
Usually treated with a combination of chemotherapy and surgery
Chemotherapy is usually given preoperatively, which allows for pathologic assessment of subsequent surgical specimen for effectiveness of drug regimen
Chemotherapy-induced necrosis ≥ 90% is considered a good response
Therapy-related cytologic changes of tumor cells are uncommon
Changes include ganglion cell differentiation, marked enlargement, hyperchromasia, pleomorphism, and presence of rhabdoid-like cells
Radiotherapy is reserved for tumors located in surgically inaccessible sites, for surgical beds in which tumors have been excised with inadequate margins, and for palliation
Prognosis
Effective chemotherapy has improved prognosis from 5-15% to 75% 5-year survival
At least 50% are long-term cures
Important factors influencing prognosis include stage of disease at time of diagnosis and site of tumor
Other important factors
Type of translocation, percentage of chemotherapyrelated tumor necrosis, presence of chimeric transcripts in cells from peripheral blood or bone marrow after treatment, and development of local recurrence
IMAGE FINDINGS
Radiographic Findings
Destructive and lytic
Poorly defined margins
Can lead to significant underestimation of the intraosseous extent on plain radiography, a feature that can be clarified by MR
Permeation of cortex usually results in concentric soft tissue mass that can be very large
Extraosseous tumor frequently erodes outer cortex producing “saucerization” of bone
Displaced periosteum deposits layers of reactive bone in onion skin-like or sunburst-like fashion
Tumor diminishes in size, sometimes completely, after therapy
MR Findings
Helpful in mapping intra- and extraosseous extent of tumor and relationship of mass to important anatomic structures
Low signal intensity on T1-weighted images
High signal intensity on T2-weighted images
May be useful in assessing post-chemotherapy tumor viability
CT Findings
Large, destructive intraosseous mass with extension into soft tissues
Bone Scan
Tumor has avid uptake of radionuclides
MACROSCOPIC FEATURES
General Features
Tan-white, fish flesh-like in appearance; usually transgresses cortex and periosteum, producing soft tissue mass
Soft tissue borders may be deceptively well circumscribed by pseudocapsule
Frequently contains areas of hemorrhage and necrosis
Thin layers of reactive bone underpin raised periosteum
Underlying osseous structure is eroded and destroyed
Intramedullary margins of tumor poorly defined
MICROSCOPIC PATHOLOGY
Histologic Features
Tumor cells are 1-2x size of lymphocytes with scant cytoplasm
Tumor cells grow in sheets or irregular islands delineated by dense fibrous tissue
Architectural features suggestive of neural differentiation traditionally include organoid growth pattern within which Homer Wright rosettes may be present
Cells have oval nuclei with finely distributed chromatin and inconspicuous nucleoli, as well as scant eosinophilic or clear cytoplasm
Cell nuclei in tumors with obvious neural differentiation are frequently slightly elongated and darkly staining
Occasionally, tumor cells are larger and have irregular nuclear contours and prominent nucleoli
Neoplasms with this morphology previously designated as atypical or large cell variant of Ewing sarcoma
Islands of tumor cells may show palisading and cording, known as adamantinoma-like variant
Mitoses may be infrequent or numerous
Necrosis may be extensive
Stroma is scant, lymphoid infiltrate absent, and supportive vascular tree is inconspicuous
Special histochemical stains (PAS) demonstrate glycogen in up to 75% of tumors
Ultrastructural Features
Primitive in appearance
Contain few organelles and may have abundant cytoplasmic glycogen
Small number of mitochondria, poorly developed Golgi complexes, free ribosomes, and inconspicuous rough endoplasmic reticulum present
Neural differentiation suggested by interdigitating cytoplasmic cell processes connected by primitive junctions, scattered microtubules, and neurosecretory granules
Definite tonofibrils and well-developed basal lamina can be identified in some keratin-positive tumors
ANCILLARY TESTS
Immunohistochemistry
Cells express vimentin, CD99 (90%), and FLI-1 (90%)
CD99 is also expressed by many other malignant small round cell tumors, including lymphoblastic lymphoma, small cell osteosarcoma, mesenchymal chondrosarcoma, and small cell carcinoma
Small cell osteosarcoma and mesenchymal chondrosarcoma are negative for FLI1
Negative staining for CD99 stain should raise possibility that tumor may not be EWS/PNET
Tumors with neuroectodermal differentiation express 1 or more neural markers including NSE, CD57, or S100
Neurofilament and keratin positivity is seen in approximately 20% of cases
Molecular Pathology
Approximately 85% harbor t(11;22)(q24;q12)
5-10% have t(21;22)(q22;q12)
< 1% of tumors show EWS fusion with ETV1 (7p22), E1AF (17q12) or FEV (2q33)
Fusion genes generated from translocations play vital role in molecular genesis of neoplasms
Translocation results in chimeric proteins that function as aberrant transcription factors governing biological behavior
Some EWS negative tumors have been shown to harbor FUS/ERG fusion – t(16;21)
If the tumor is also FUS/ERG fusion negative, then possibility of CIC/DUX4 fusion should be considered
DIFFERENTIAL DIAGNOSIS
Malignant Lymphoma
Cells in large cell lymphoma are frequently larger than those of EWS/PNET
Cells in large cell lymphoma also have more cytoplasm, and their nuclei may be irregular, cleaved, and hyperlobated
Lymphoblastic lymphoma is composed of uniform round cells and, as with most lymphomas, frequently contains admixed benign infiltrating lymphocytes
Can express CD99 but are FLI-1 negative and TdT positive
Lymphomas express lymphoid antigens; ultrastructurally, cells have marginated chromatin, lack intercellular junctions, and do not contain glycogen
Chromosomal translocations associated with malignant lymphoma do not include those identified in Ewing sarcoma/PNET
Neuroblastoma
Metastatic undifferentiated neuroblastoma can be difficult to distinguish from EWS/PNET on strict histologic grounds
Neuroblastoma negative for CD99 and FLI-1; N-MYC is amplified
Helpful findings in poorly differentiated neuroblastoma are presence of neuropils and evidence of ganglion cell differentiation
Neuroblastoma has loss of chromosome 1p and does not have a translocation involving chromosome 22
Small Cell Osteosarcoma
If matrix is prominent, EWS/PNET is easily distinguished from small cell osteosarcoma
EWS may contain reactive woven bone that can cause confusion with osteosarcoma; look for osteoblastic rimming
Mesenchymal Chondrosarcoma
Contains areas of chondroid differentiation admixed with malignant small round cell component
Tumor has nodular architecture and prominent vascular tree that frequently has hemangiopericytomalike pattern, which is usually not present in EWS/PNET
DIAGNOSTIC CHECKLIST
Pathologic Interpretation Pearls
Sheets of malignant small round cells: Think EWS/PNET
SELECTED REFERENCES
1. Machado I et al: Molecular diagnosis of Ewing sarcoma family of tumors: a comparative analysis of 560 cases with FISH and RT-PCR. Diagn Mol Pathol. 18(4):189-99, 2009
2. Shing DC et al: FUS/ERG gene fusions in Ewing’s tumors. Cancer Res. 63(15):4568-76, 2003
Image Gallery
Imaging Features
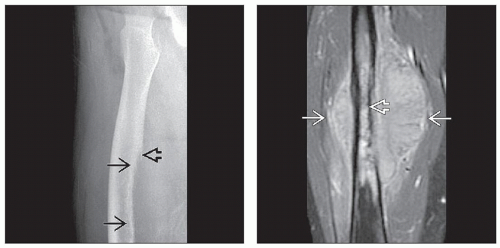 (Left) Radiograph of EWS shows a large saucerization defect along the femoral shaft  causing thinning of the cortex. This results from the tumor permeating the cortex, forming an extraosseous soft tissue mass. A Codman triangle is seen proximally causing thinning of the cortex. This results from the tumor permeating the cortex, forming an extraosseous soft tissue mass. A Codman triangle is seen proximally  . (Right) T1-weighted, contrast-enhanced coronal MR with fat saturation shows a Ewing sarcoma arising in the diaphysis of the femur and extending into adjacent soft tissues . (Right) T1-weighted, contrast-enhanced coronal MR with fat saturation shows a Ewing sarcoma arising in the diaphysis of the femur and extending into adjacent soft tissues  , causing saucerization of the medial femoral shaft , causing saucerization of the medial femoral shaft 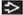 . . |
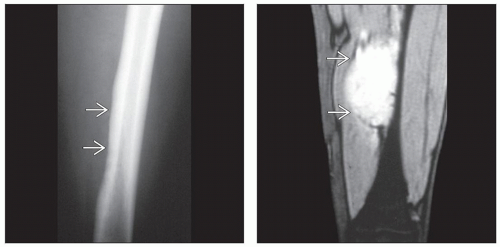 (Left) Radiograph shows Ewing sarcoma arising in the diaphysis of the femur, producing a slight increase in density within the medullary cavity. The tumor has extended into adjacent soft tissues, causing secondary subtle saucerization defect  . (Right) MR shows Ewing sarcoma arising in the shaft of the mid diaphysis of the femur, forming a large, hyperintense mass in the adjacent lateral soft tissues . (Right) MR shows Ewing sarcoma arising in the shaft of the mid diaphysis of the femur, forming a large, hyperintense mass in the adjacent lateral soft tissues  . The intramedullary component is barely visible in this image. . The intramedullary component is barely visible in this image. |
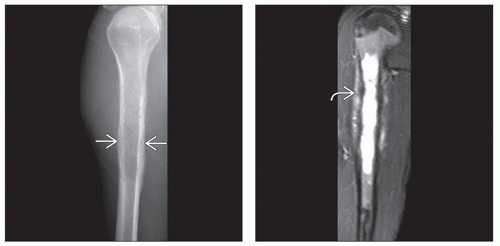 (Left) X-ray of a Ewing sarcoma arising in the proximal humerus shows a lucent lesion with areas of endosteal resorption and periosteal new bone formation
 along the medial and lateral cortices. (Right) Coronal T2-weighted MR of a Ewing sarcoma shows hyperintense signal replacing the marrow in the proximal humeral shaft with permeation of tumor through an irregular thinned cortex. The linear dark signal perpendicular to the cortex along the medial and lateral cortices. (Right) Coronal T2-weighted MR of a Ewing sarcoma shows hyperintense signal replacing the marrow in the proximal humeral shaft with permeation of tumor through an irregular thinned cortex. The linear dark signal perpendicular to the cortex  represents sunburst periosteal new bone. represents sunburst periosteal new bone.Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|