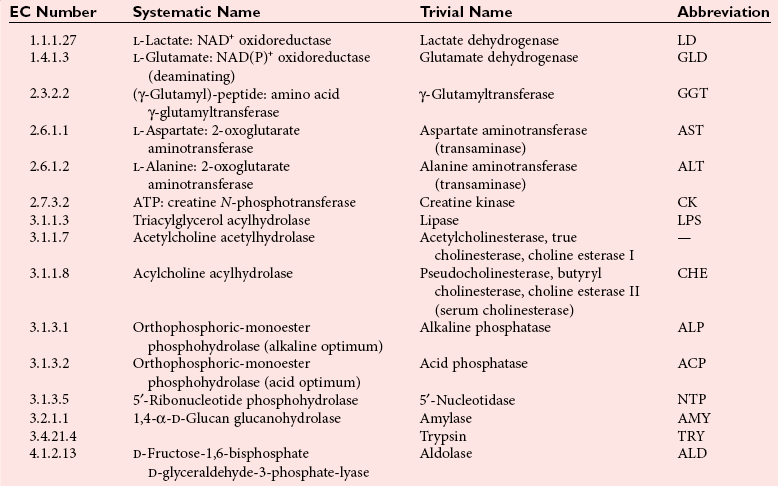
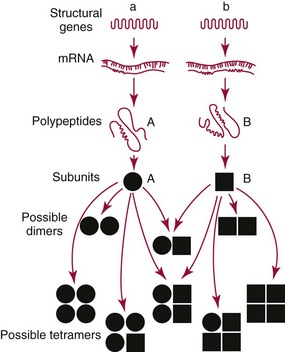
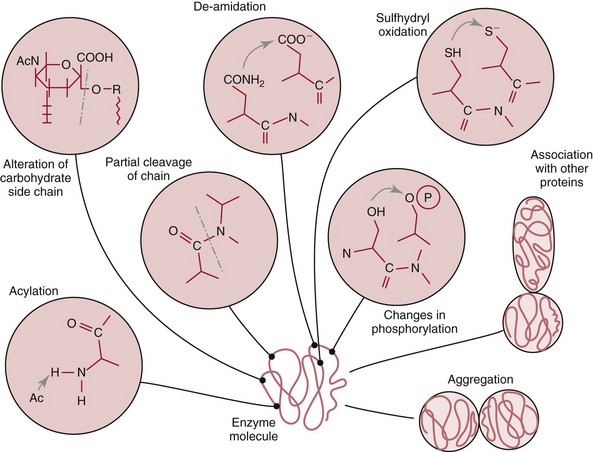
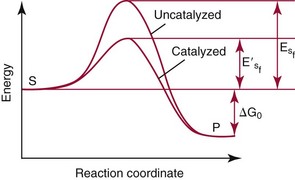
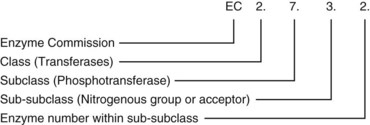
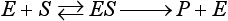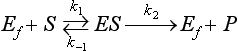Chapter 15 Because this combination of trivial common names and semisystematic names was found to be inadequate, in 1955 the International Union of Biochemistry (IUB) appointed an Enzyme Commission (EC) to study the problem of enzyme nomenclature. Its subsequent recommendations, with periodic updating, provide a rational and practical basis for identifying all enzymes now known and enzymes that will be discovered in the future (http://www.chem.qmw.ac.uk/iubmb/enzyme/accessed May 6, 2011).11 Table 15-1 lists some selected enzymes of clinical interest, identified by trivial, abbreviated, and systematic names and by their code numbers. Although it is not recommended by the EC, it is a common and convenient practice to use capital letter abbreviations for the names of certain enzymes, such as ALT (formerly GPT) for alanine aminotransferase. Other examples are AST for aspartate aminotransferase, LD for lactate dehydrogenase, and CK for creatine kinase (see Table 15-1). All enzyme molecules possess the primary, secondary, and tertiary structural characteristics of proteins (see Chapter 21). In addition, most enzymes exhibit quaternary structure. The primary structure, the linear sequence of amino acids linked through their α-carboxyl and α-amino groups by peptide bonds, is specific for each type of enzyme molecule. Each polypeptide chain is coiled up into three-dimensional secondary and tertiary structure. Secondary structure refers to the conformation of limited segments of the polypeptide chain, namely α-helices, β-pleated sheets, random coils, and β-turns. The arrangement of secondary structural elements and amino acid side chain interactions that define the three-dimensional structure of the folded protein is referred to as its tertiary structure. In many cases, biological activity, such as the catalytic activity of enzymes, requires two or more folded polypeptide chains (subunits) to associate to form a functional molecule. The arrangement of these subunits defines the quaternary structure. The subunits may be copies of the same polypeptide chain (e.g., the MM isoenzyme of creatine kinase, the H4 isoenzyme of lactate dehydrogenase), or they may represent distinct polypeptides. The biological activity of a protein molecule depends generally on the integrity of its structure, and any disruption of its structure is accompanied by loss of activity, a process known as denaturation. If the process of denaturation is minimal, it may be reversed with the recovery of enzyme activity upon removal of the denaturing agent. However, prolonged or severe denaturing conditions result in an irreversible loss of activity. Denaturing conditions include elevated temperatures, extremes of pH, and chemical addition. Heat inactivation of most enzymes takes place at an appreciable rate at room temperature and in most cases becomes almost instantaneous above 60 °C. The polymerases are an exception and retain activity at temperatures as high as 90 °C—a property that has been made use of in the polymerase chain reaction (see Chapter 17). Low temperatures are used to preserve enzyme activity, especially in aqueous solutions, such as serum (see Chapter 22). Extremes of pH also cause unfolding of enzyme molecular structures and, except for a few exceptions, should be avoided when enzyme samples are preserved. Addition of chemicals, such as urea and detergents, disrupts hydrogen bonds and hydrophobic interactions so that exposure of enzymes to strong solutions of these reagents results in inactivation. With the exception of enzymes such as proteases, nucleases, and amylases, which act on macromolecular substrates, enzyme molecules are considerably larger than the molecules of their substrates. Consideration of the structure of an enzyme’s active site and its relationship to the structures of the enzyme’s substrate(s) in its ground and transition states is necessary to understand the rate enhancement and specificity of the chemical reactions performed by the enzyme. The active site of an enzyme will vary between enzymes but in general,5 1. The active site of an enzyme is relatively small compared with the total volume of the enzyme molecule because its structure may involve less than 5% of the total amino acids in the molecule. 2. The active sites of enzymes are three-dimensional structures that are formed as a result of the overall tertiary structure of the protein. This results when the amino acids and cofactors in the active site of an enzyme are spatially structured in an exact, three-dimensional relationship with respect to one another and the structure of the substrate molecule. 3. Typically, the attraction between the molecules of the enzyme and its substrate molecules is noncovalent binding. Physical forces used in this type of binding include hydrogen bonding, electrostatic and hydrophobic interactions, and van der Waals forces. 4. Active sites of enzymes typically occur in clefts and crevices in the protein. This excludes bulk solvent and reduces the catalytic activity of the enzyme. 5. The specificity of substrate binding is a function of the exact special arrangement of atoms in the enzyme active site that complements the structure of the substrate molecule. An isoenzyme is defined as “one of a group of related enzymes catalyzing the same reaction but having different molecular structures and characterized by varying physical, biochemical, and immunologic properties.” However, the IUB recommends that use of the term isoenzyme be restricted to forms that originate at the level of the genes that encode the structures of the enzyme proteins in question.4 The number of allelic variants and the frequency with which particular variants occur within the population vary considerably from one enzyme to another. For example, mutations at either of the two principal loci that determine human lactate dehydrogenase are extremely rare, but a high incidence of mutant alleles occurs at the single locus that determines the structure of placental alkaline phosphatase. More than 400 mutations in the glucose-6-phosphate dehydrogenase gene have now been identified on the X chromosome [up-to-date genetic information on this and other enzymes can be obtained from the Online Mendelian Inheritance in Man (OMIM) database at http://www.ncbi.nlm.nih.gov/Omim/searchomim.html/ accessed May 6, 2011]. Some of these alleles are extremely rare, whereas others occur with appreciable frequency in particular populations or geographical locations. When isoenzymes, because of variation at a single locus, occur with appreciable frequency in a human population, the population is said to be polymorphic with respect to the isoenzymes in question. The number of different hybrid isoenzymes that can be formed from two nonidentical protomers depends on the number of subunits in the complete enzyme molecule. For a dimeric enzyme, one mixed dimer (hybrid isoenzyme) can be formed. If the enzyme is a tetramer, three heteropolymeric isoenzymes may be formed (Figure 15-1). Examples of hybrid isoenzymes include the mixed MB dimer of creatine kinase and the three hybrid isoenzymes—LD-2, LD-3, and LD-4—of lactate dehydrogenase. Post-translational modifications of enzyme molecules give rise to multiple forms known as isoforms (Figure 15-2). Serum isoforms of creatine kinase are formed as part of the normal clearance process of the cell. Human myocardial and skeletal muscle tissues have the CK-MM and CK-MB isoenzymes, which are modified upon release into the circulation. This modification is due to sequential removal of the C-terminal amino acid, lysine, by the action of carboxypeptidase (see Chapter 22 for additional details). Modifications affecting nonprotein components of enzyme molecules may also lead to molecular heterogeneity. Many enzymes are glycoproteins, and variations in carbohydrate side chains are a common cause of nonhomogeneity of preparations of these enzymes. Some carbohydrate moieties, notably N-acetylneuraminic acid (sialic acid), are strongly ionized and consequently have a profound effect on some properties of enzyme molecules.1 For example, removal of terminal sialic acid groups from human liver and/or bone alkaline phosphatase with neuraminidase greatly reduces the electrophoretic heterogeneity of the enzyme. Structural differences between the multiple forms of an enzyme give rise to greater or lesser differences in physicochemical properties, such as electrophoretic mobility, resistance to inactivation, and solubility, or in catalytic characteristics, such as the ratio of reaction with substrate analogs or response to inhibitors. Methods of isoenzyme analysis have therefore been designed to investigate a wide range of catalytic and structural properties of enzyme molecules.9 However, it is usually possible to make only limited deductions about the nature of the underlying structural differences between isoenzymes that are responsible for the dissimilar properties. Equally, the changes in catalytic and other properties that may result from specific structural alterations in enzyme molecules are difficult to predict from current theoretical knowledge of the relationship between structure and function of proteins.22 The International System of Units (SI)-derived unit for catalytic activity (see Chapter 9) is the katal, defined as moles converted per second. The name katal had been used for this unit for decades but did not become an official SI-derived unit until 1999 with Resolution 12 of the 21st French Conférence Général des Poids et Mesures (CGPM), on the recommendation of the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC). Both the International Union of Pure and Applied Chemistry (IUPAC) and the IUB now recommend that enzyme activity be expressed in moles per second, and that the enzyme concentration be expressed in terms of katals per liter (kat/L).8 Thus, 1 U = 10−6 mol/60 s = 16.7 × 10−9 mol/s, or 1.0 nkat/L = 0.06 U/L. Enzymes act through the formation of an enzyme–substrate (ES) complex, in which a molecule of substrate is bound to the active center of the enzyme molecule. The binding process transforms the substrate molecule to its activated state. The energy required for this transformation is provided by the free energy of binding of S to E. Therefore, activation takes place without the addition of external energy, so that the energy barrier to the reaction is lowered and the breakdown to products is accelerated (Figure 15-3). The ES complex breaks down to give the reaction products (P) and free enzyme (E):
Enzyme and Rate Analyses
Basic Principles
Enzyme Nomenclature
Enzymes as Proteins
Specificity and the Active Center
Isoenzymes and Other Multiple Forms of Enzymes
Genetic Origins of Enzyme Variants
Nongenetic Causes of Multiple Forms of Enzymes
Differences in Properties Between Multiple Forms of Enzymes
Units for Expressing Enzyme Activity
Enzyme Kinetics
Factors Governing the Rate of Enzyme-Catalyzed Reactions
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Enzyme and Rate Analyses