Enteroviruses: Polioviruses, Coxsackieviruses, Echoviruses, and Newer Enteroviruses
Mark A. Pallansch
M. Steven Oberste
J. Lindsay Whitton
History
The history of enteroviruses (EVs) is very much the history of poliovirus (PV). In fact, many of the PV milestones are landmarks in the study of EV and, in fact, all of virology.
Poliomyelitis is believed to be an ancient disease. It has been suggested that the depiction of a young man with an atrophic limb on an Egyptian stele from the second millennium BC represents a sequela of poliomyelitis.197 The first clinical descriptions of poliomyelitis were made in the 1800s, with reports of cases of paralysis with fever. In 1840, von Heine153 published a monograph more specifically describing the affliction. His contributions and those published later by Medin271 from Sweden led to paralytic poliomyelitis being referred to as Heine-Medin disease. Another early report, by Charcot and Joffroy,65 described the pathologic changes in the anterior horn motor neurons of the spinal cord in poliomyelitis.
The 1900s began a new era in poliomyelitis investigations and the beginning of an understanding of the infectious nature of this disease. Wickman418 and others recognized the communicable nature of poliomyelitis, the importance of asymptomatic infected individuals in transmission of PV, and the role of enteric infection in disease pathogenesis. The role of the gastrointestinal tract in the initiation and spread of PV infection was later confirmed by Trask et al.396 In a classic study, Viennese investigators Landsteiner and Popper241 proved the infectious nature of poliomyelitis by successfully transmitting the clinical disease and its pathology to monkeys following inoculation of central nervous system (CNS) tissue homogenates from human cases.
Despite this progress, a number of unfortunate misconceptions emerged about poliomyelitis that initially confused scientists and misdirected efforts for control. These misconceptions included a belief that the virus was exclusively neurotropic, that
the nasopharynx was a major site for virus entry into the CNS, and that the virus spread to the nervous system before viremia and by way of the olfactory nerve. As a result of these misconceptions and the failure of several poorly conceived immunization attempts, some with rather disastrous results,328 an atmosphere of pessimism existed by the middle of the 20th century concerning the eventual control of poliomyelitis, even among scientists working in the field. In 1945, Burnet49 wrote, “The practical problem of preventing infantile paralysis has not been solved. It is even doubtful whether it ever will be solved.” The eventual realization that virus entered via the oral–gastrointestinal route and that CNS disease followed a viremia did much to boost hopes for effective immunization.36
the nasopharynx was a major site for virus entry into the CNS, and that the virus spread to the nervous system before viremia and by way of the olfactory nerve. As a result of these misconceptions and the failure of several poorly conceived immunization attempts, some with rather disastrous results,328 an atmosphere of pessimism existed by the middle of the 20th century concerning the eventual control of poliomyelitis, even among scientists working in the field. In 1945, Burnet49 wrote, “The practical problem of preventing infantile paralysis has not been solved. It is even doubtful whether it ever will be solved.” The eventual realization that virus entered via the oral–gastrointestinal route and that CNS disease followed a viremia did much to boost hopes for effective immunization.36
Building on studies of others, Enders et al.105 performed a landmark study showing that PV could be propagated in nonneural tissue culture. These investigations had implications for all of virology because they indicated, first, that PV grew in various tissue culture cells that did not correspond to the tissues infected during the human disease, and second, that PV destroyed cells with a specific cytopathic effect. Neutralization tests showed that PV has three serotypes,39 and serologic tests25 confirmed that most infected individuals do not manifest clinical disease. These investigations laid a critical framework for the development of a vaccine, and they clarified a host of confusing data, such as the apparent presence of second attacks of poliomyelitis.
A variety of vaccines were subsequently produced, with the most well known being the Salk inactivated polio vaccine (IPV) delivered via the intramuscular route (licensed in 1955 in the United States) and the Sabin live, attenuated vaccine (oral polio vaccine [OPV]) delivered via the oral route (licensed in 1961–1962). The importance of these vaccines and the individuals who produced them can begin to be realized by noting that more Americans knew the name of Jonas Salk than the president of the United States. The real impact of these vaccines will ultimately be felt with the complete global eradication of poliomyelitis. The eradication will undoubtedly provide a fitting dramatic finale to the compelling story of poliomyelitis.
Poliovirus work has had a continuing significant impact on the field of molecular virology. PV was the first animal virus completely cloned and sequenced,227,337 the first RNA animal virus for which an infectious clone was constructed,336 and the first human virus that had its three-dimensional structure solved by x-ray crystallography.164 In 1989, Mendelsohn et al.279 identified the PV receptor, CD155, a finding that was followed by the generation of mice carrying CD155 as a transgene.234,344
Coxsackieviruses (group A) were first isolated during poliomyelitis outbreaks in 1947 from the feces of paralyzed children in Coxsackie, New York.89 These isolates were obtained by inoculation of suckling mice, the pathogenicity in mice clearly differentiating these viruses from PV. In the following year, the first coxsackievirus (CV) group B was isolated from cases of aseptic meningitis.276 The original CV group A (CVA) isolates produced myositis with flaccid hind limb paralysis in newborn mice, whereas the coxsackieviruses group B (CVB) produced a spastic paralysis and generalized infection in newborn mice, with myositis and involvement of the brain, pancreas, heart, and brown fat.
In 1951, echoviruses were first isolated from the stool of asymptomatic individuals.349 Echoviruses received their name because they were enteric isolates, cytopathogenic in tissue culture, isolated from humans, and orphans (i.e., unassociated with a known clinical disease). Subsequent studies have shown that echoviruses, in fact, do cause a variety of human diseases. After this period of rapid growth in the number of enteroviruses, there were several decades where new enteroviruses were uncommonly identified. This changed with the introduction of molecular detection methods, and the last 15 years have seen a rapid expansion in the number of recognized enteroviruses. This period of discovery is still in progress.
Infectious Agents
Physical and Chemical Properties
Enteroviruses are distinguished from other picornaviruses on the basis of physical properties, such as buoyant density in cesium chloride and stability in weak acid. Many aspects of enteroviral pathology, transmission, and general epidemiology are directly related to the biophysical properties and their cytolytic life cycle. The infectious virus is relatively resistant to many common laboratory disinfectants, including 70% ethanol, isopropanol, dilute Lysol, and quaternary ammonium compounds. The virus is insensitive to lipid solvents, including ether and chloroform, and it is stable in many detergents at ambient temperature. Formaldehyde, glutaraldehyde, strong acid, sodium hypochlorite, and free residual chlorine inactivate enteroviruses. Concentration, pH, extraneous organic materials, and contact time affect the degree of inactivation by these compounds. Similar inactivation is achieved when virus is present on fomites, although conditions may not be exactly comparable.1 In general, most reagents that inactivate EV depend on active chemical modification of the virion, whereas most extractive solvents have no effect.
Enteroviruses are relatively thermostable, but less so than hepatitis A virus. Most enteroviruses are readily inactivated at 42°C, although some sulfhydryl reducing agents and magnesium cations can stabilize viruses so that they are relatively stable at 50°C.8,99 The relative sensitivity to modest elevations in temperature makes it possible to use pasteurization to inactivate EV in many biologically active preparations.160
As with other infectious agents, ultraviolet light can be used to inactivate EV, particularly on surfaces. In addition, the process of drying on surfaces significantly reduces virus titers. The degree of virus loss by drying is related to porosity of the surfaces and the presence of organic material.2 Many studies of EV inactivation have been conducted using PV as a model enterovirus. A report describing strain-specific differences for glutaraldehyde inactivation among echovirus 25 isolates implies, however, that the assumption that PV is representative of all EV may not be valid.62 The inactivation of infectivity may not be directly related to the destruction of the viral genome, because the polymerase chain reaction (PCR) can be used to amplify viral RNA, even after inactivation of virus has occurred.259 This would suggest that reactivation of infectivity may be possible in some circumstances. In fact, some examples of recovered infectivity have been reported through increased multiplicity of infection in cell culture,372,443 but the practical significance of these observations is not clear.
Antigenic Characteristics and Taxonomy
As described in Chapter 16, the picornaviruses are among the simplest RNA viruses, having a highly structured capsid with little place for elaboration. Yet, despite the limited
genetic material and structural constraints, evolution within the picornaviruses has resulted in a large number of readily distinguishable members. This variability has been categorized antigenically as serotype.
genetic material and structural constraints, evolution within the picornaviruses has resulted in a large number of readily distinguishable members. This variability has been categorized antigenically as serotype.
Each of the serotypes correlates with the immunologic response of the human host, protection from disease, receptor usage, and, to a lesser extent, the spectrum of clinical disease. These correlations, however, have only a partial relationship with the original classification of enteroviruses into polioviruses, coxsackie A or B viruses, and echoviruses, based on biological activity and disease: human CNS disease with flaccid paralysis (poliomyelitis); flaccid paralysis in newborn mice, human CNS disease, and herpangina (coxsackie A viruses); spastic paralysis in newborn mice and human CNS and cardiac disease (coxsackie B viruses); and no disease in mice and (originally) no human disease (echoviruses). Within each of these groups, isolates can be readily distinguished on the basis of antigenicity as measured with antisera raised in animals. The original classification scheme broke down with the identification of viruses serologically identical to known echoviruses that were found to cause disease in mice and humans. This and other inconsistencies led to a numbering of new EV serotypes starting with EV68. These antigenic groupings, which define the serotypes, became increasingly more complicated as the number of different viruses grew. Despite these limitations, the serotype remains the single most important physical and immunologic property that distinguishes the different EVs. Most of the prototype EV strains are maintained in the American Type Culture Collection, Manassas, Virginia, and in many of the World Health Organization (WHO) collaborating reference laboratories.
Despite the importance of the antigenic properties, the introduction of molecular typing methods and a reassessment of the limitations of the old classification scheme led to the development of the current classification system that divides the members of the EV genus into species on the basis of genome organization and sequence similarity as well as biological properties (Table 17.1).
The human enteroviruses are now classified into four species: Enterovirus A (EV-A), EV-B, EV-C, and EV-D. In this system, members within an EV species:
share greater than 70% aa [amino acid] identity in the polyprotein, share greater than 60% aa identity in P1, share greater than 70% aa identity in the nonstructural proteins 2C + 3CD, share a limited range of host cell receptors, share a limited natural host range, have a genome base composition (G + C) which varies by no more than 2.5%, share a significant degree of compatibility in proteolytic processing, replication, encapsidation, and genetic recombination.230
Coding for the capsid proteins, the P1 region provides a reliable correlation between sequence relatedness and the traditional definition of serotype. This also appears to be true for the various individual capsid protein regions, with the exception of VP4; the VP4 sequence does not always correlate with serotype and, therefore, is not reliable for serotype identification.
The molecular studies have also provided a framework in which the EV antigenic relationships can be better understood. These studies suggest that the nucleotide sequence of VP1 can function as an excellent surrogate for the reference antigenic typing methods that use neutralization tests in order to distinguish EV serotypes. VP1 nucleotide sequence identity of at least 75% (85% aa identity) between an isolate and a serotype prototype strain suggests that the isolate is serotypically identical to the prototype (assuming that the next highest identity with other prototype strains is less than 70%). For example, a capsid sequence identity of 85.4% aa between CVA3 and A8 compared with a mean sequence identity among prototype strains of 71.5% confirmed the antigenic relationships that had previously been described and suggested that these two viruses probably derived relatively recently from a common ancestor.318 Similarly, capsid sequences with more than 96% identity confirmed the antigenic relationships between CVA11 and A15 and between A13 and A18;46 as a result, CVA15 and A18 have been reclassified as A11 and A15, respectively. In this way, the use of VP1 sequencing studies has supported and clarified early serologic data and also led to proposals regarding the classification of isolates into new EV serotypes.312,316
No general correlation is found, however, between sequence similarities with serotype in genome segments outside of the capsid region because of frequent recombination in the noncapsid regions. For example, sequencing studies have shown that phylogenetic trees constructed from sequences from varying genome regions of members of EV-C and Poliovirus have incongruities between the capsid region and noncapsid regions,46 suggesting that viruses with a PV capsid may recombine with these CV nonstructural protein coding regions to acquire different nonstructural protein sequences and, similarly, viruses with a CV capsid protein sequence may recombine to acquire different nonstructural protein sequences.255 These findings imply that recombination occurs between PV and other EV-C viruses within the nonstructural protein coding regions, and this shuffling of different nonstructural protein coding regions may lead to serotypes with selective advantages that become dominant. The frequency of recombination in the noncapsid region supports the idea that serotype is defined by the capsid region and that limited correlations likely exist between the serotype of isolates and other phenotypic characteristics not associated with the capsid proteins. The findings also demonstrated that the phylogenetic clustering of prototype strains changes, depending on the nonstructural region that is analyzed.
Recombination within the nonstructural proteins was also found among EV-A and EV-B prototype members with other members of the same species, consistent with their classification into two separate species.314,318 An analysis of multiple isolates within several EV-B serotypes319 found relatively frequent interserotypic recombination of the noncoding regions, which appeared to occur at least once every 6 years for the isolates that were analyzed. Although no evidence was found of interserotypic recombination within the capsid (perhaps because of structural constraints specific for a particular serotype), intraserotypic recombination within this region appeared to occur.
Additional sequencing studies showed that the 5′ untranslated region (UTR) of enteroviruses forms two clusters: the viruses of EV-C and EV-D compose cluster I, whereas EV-A and EV-B compose cluster II.46,188
Sequencing studies have also demonstrated significant similarities between human rhinoviruses and the human enteroviruses, resulting in a reclassification of the human rhinoviruses as members of three different species within the genus Enterovirus.230 Despite the reclassification, the rhinoviruses have several unique properties and are covered separately in this volume (see Chapter 18).
Table 17.1 Picornavirus Genera, Species,a and (Sero)Types | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In addition to the genetic relatedness, many different EV serotypes share some antigenicity. For example, PV1 and 2 share a common antigen, and antigenic relationships also exist between coxsackieviruses A3 and A8, A16 and EV71, and A24 and EV70, and between echoviruses 6 and 30, and 12 and 29. When virions are disrupted by heating, particularly in the presence of detergent, nonsurface antigens are exposed that are shared broadly among many EVs.280
Despite this lack of understanding of molecular variation in virus structure as measured by polyclonal antibodies, high-resolution studies of the virion surface have been particularly useful in identifying the targets of neutralization of EV by monoclonal antibodies.165,327 Other less investigated antigenic sites elicit immune responses that are not neutralizing but nevertheless contribute to serotype identity. The observed structure of some antigenic sites has been shown to span noncontinuous polypeptide chains, providing an explanation for why antigenicity of the virus is destroyed by disruption of the virion structure.
Antisera raised in animals to each of the enteroviruses are largely type specific and are used to determine serotype in a neutralization assay. The PV neutralizing antibody response is serotype specific, with the exception of some minor cross-reaction between PV1 and 2. A monoclonal antibody has been described that reacts with this shared site that is not found on PV3.403 As noted, heat-disrupted virions, particularly those heated in the presence of detergent, induce antibodies that react with many EVs.280 These broadly reacting antibodies are generally not neutralizing, and at least one of these epitopes has been mapped to the amino-terminal region of capsid protein VP1.362 Although measurable in vitro differences are found in antigenic properties among strains within a serotype, the significance of these differences during natural infection has not been determined. Several PVs isolated during outbreaks have demonstrated different antigenic properties when compared with the reference vaccine strains.140,186 In all cases, immunity derived from vaccination has been sufficient to provide protection and control of these strains.43 In addition, even in the face of massive immunization campaigns, no antigenic escape mutants resistant to neutralization have ever been observed, and successive genotypes of PV have been eliminated. Natural antigenic variants have also been identified with panels of monoclonal antibodies for several nonpolio EVs.148,329
Propagation and Assay in Cell Culture
One of the prominent characteristics of enteroviruses is the cytolytic nature of growth in cell culture. For many years, PV was the prototype of a lytic viral infection. At the microscopic level, infection is usually manifest within 1 to 7 days by the appearance of a characteristic cytopathic effect, which features visible rounding, shrinking, nuclear pyknosis, refractility, and cell degeneration (Fig. 17.1). The earliest effects can be seen in less than 24 hours if the inoculum contains many infectious particles. With fewer virions, however, visible changes are not recognizable for several days, although a sufficient number of cells are infected. In addition, some EVs either do not cause cytopathic effect at all or do so only after several passages. In general, once focal cytopathic effect is detected, infection spreads rapidly throughout the cell sheet with total destruction of the monolayer, sometimes in a matter of hours.
All known EVs can be propagated in either cell culture or in suckling mice. Most of the serotypes can be grown in at least one human or primate continuous cell culture. No cell line, however, can support the growth of all cultivable EVs. Even after many years of experimentation, a few serotypes (e.g., CVA19) can be propagated only in suckling mice. The typical host range of human EV in cell cultures or animals is shown in a broad, generalized way in Table 17.2 and is not clearly associated specifically with a given virus species.275 Infection of target cells depends on viruses binding to specific receptors on the cell surface. Collectively, the EVs use many different receptors. A practical adaptation resulting from the identification and genetic cloning of EV receptors is the introduction of the receptor into animals and cells that do not normally permit virus infection. This approach has resulted in advances in understanding both the pathogenesis of PV infection in a nonprimate animal model system and its practical application in the diagnostic laboratory. The L20B cells, which are murine cells that express CD155, are now used routinely to selectively isolate PV (see Diagnosis) as part of the global PV laboratory network supporting the poliomyelitis eradication initiative.333
Infection in Experimental Animals: Host Range
The natural host for all human enteroviruses is the human. Although serologically distinct picornaviruses with the same physical properties as human EV have been found in many animals, human beings do not usually have recognizable infections with these animal EV. On the other hand, some animals are susceptible to experimental infection with human EV. These include nonhuman primates and CD155 transgenic mice for polioviruses, mice and some monkeys for coxsackieviruses A and B, and monkeys for echoviruses. Human EV can infect nonhuman primates, perhaps related to the homology that several simian EVs share with human viruses, but the infections appear to be largely subclinical.313,334 Among higher primates, chimpanzees and gorillas appear to be able to acquire PV infection and develop disease from humans through natural exposure.100 CVB5 is closely related antigenically to the porcine EV causing swine vesicular disease, with about 50% genetic homology over the entire genome. Genetic studies of a number of strains of swine vesicular disease virus, as well as epidemiologic information gleaned from outbreaks, strongly suggest that a human CVB5 was specifically introduced into swine decades ago and led to establishment in this new host.448
Although most coxsackie A viruses have been successfully grown in various cell culture systems, isolation from clinical specimens is sometimes unsuccessful, necessitating the inoculation of suckling mice. Inoculation of suckling mice and subsequent virus identification is a process analogous to that of cell culture inoculation. A blind passage in mice may be necessary if the inoculum is of very low titer or, possibly, because passage of the virus is needed for it to adapt to growth in mice. The two groups of coxsackieviruses can be distinguished by the distinct pathology that they cause in mice (Figs. 17.2 and 17.3). With CVA infection, newborn mice develop flaccid paralysis and severe, extensive degeneration of skeletal muscle (sparing the tongue, heart, and CNS), and they may have renal lesions. Death usually occurs within a week. CVB infection proceeds more slowly and is characterized by spastic paralysis and tremors associated with encephalomyelitis, focal myositis, necrosis of brown fat pads, myocarditis, hepatitis, and acinar cell
pancreatitis. Echoviruses, except for some isolates of echovirus type 9, do not generally cause disease in mice.
pancreatitis. Echoviruses, except for some isolates of echovirus type 9, do not generally cause disease in mice.
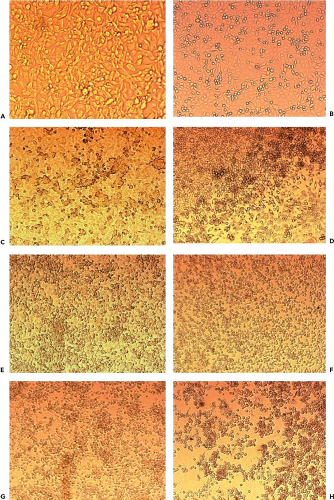 Figure 17.1. Normal and poliovirus (PV)-infected rhabdomyosarcoma (RD) cells and CD155 transgenic mouse L cells (L20B) in culture. A: Monolayer of normal RD cells in culture. B: RD cell culture showing early stage of cytopathic effect typical of PV infection. Approximately 25% of the cells in the culture show cytopathic effect (especially rounding) indicative of virus multiplication (1+ cytopathic effect score). C: RD cell culture illustrating more advanced cytopathic effect (3+ to 4+ cytopathic effect). D: Almost 100% of the cells are affected, and most of the cell sheet has come loose from the wall of the culture tube. E–H: Similar stages of cytopathic effect are shown as in A–D, but in this case L20B cells are infected with PV. |
Table 17.2 Usual Host Range of Human Enteroviruses: Animal and Tissue Culture Spectruma | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
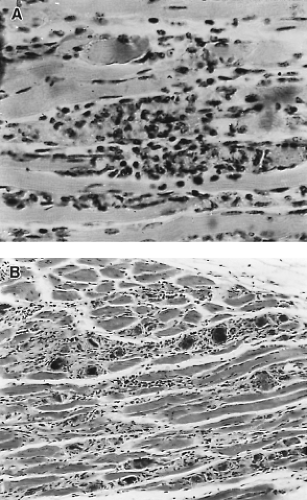 Figure 17.2. Photomicrographs of lesions of striated muscle in suckling mice infected with coxsackieviruses. A: Two sarcolemmic tubes with numerous mononuclear phagocytes and remnants of hyaline material within the sarcolemmic sheaths. Acute stage of infection with Conn-5 strain, prototype of coxsackievirus B1 (CVB1) (×450). B: Partly mineralized residual masses surrounded by fibrous capsules, and sometimes by giant cells. The remainder of the muscle is completely restored. Texas-1 strain of CVA4, 11 days after onset of paralysis (×180). (From Melnick JL. Current status of poliovirus infections. Clin Microbiol Rev 1996;9[3]:293–300, with permission.) |
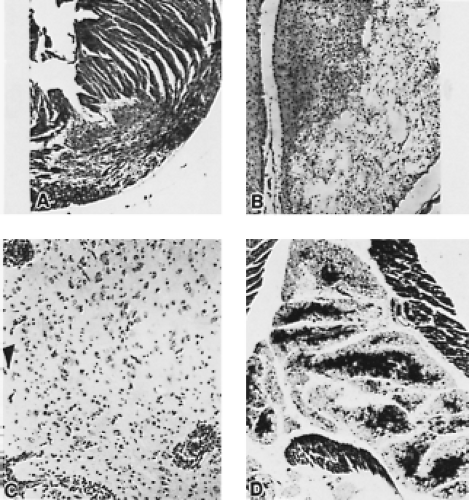 Figure 17.3. Photomicrographs of lesions in heart, brain, and fat lobules of suckling mice in acute stage of infection with Conn-5 strain of coxsackievirus B1 (CVB1) (90). A: Heart. Large zone of myocardial necrosis at apex of left ventricle (×170). B: Brain. Rarefaction after necrosis in cerebrum (×140). C: Brain. Encephalitis showing marked perivascular cuffing and leptomeningitis (×190). D: Fat lobes. Interlobular edema and acute necrotizing steatitis, illustrating the selective destruction of the peripheral parts of the lobules, which are shown as pale margins, with preservation of the central parts, which are shown as dark fuchsinophilic areas (×72). (From Melnick JL. Current status of poliovirus infections. Clin Microbiol Rev 1996;9[3]:293–300, with permission.) |
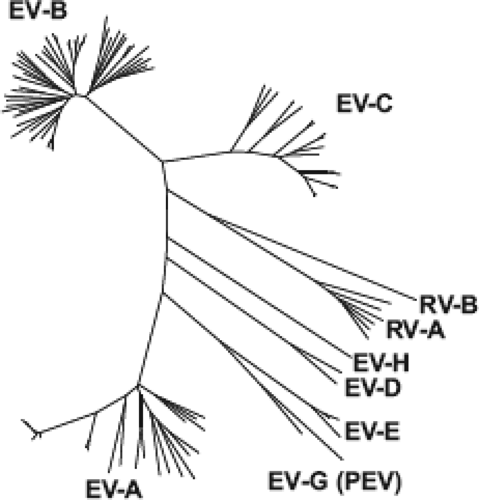 Figure 17.4. Dendrogram of genus Enterovirus. The figure illustrates the phylogenetic relationship among the prototype strains (see Tables 17.4 through 17.7) within the genus Enterovirus and the distinct grouping of isolates into each of the species of viruses that affects humans and other related animal enteroviruses based on the amino acid sequence of the P1 (capsid) coding region of the genomes. Species: EV-A (formerly Human enterovirus A); EV-B (formerly Human enterovirus B); EV-C (formerly Human enterovirus C); EV-D (formerly Human enterovirus D); RV-A (formerly Human rhinovirus A); RV-B (formerly Human rhinovirus B); EV-E (formerly Bovine enterovirus); EV-G (formerly Porcine enterovirus B); EV-H (formerly Simian enterovirus B). Not shown: EV-F (Bovine enterovirus 2), EV-J (Simian virus 6 and related viruses), and RV-C. (Data from M. S. Oberste.) |
Other Human Picornaviruses
In addition to the enteroviruses, rhinoviruses (Chapter 18), and hepatitis A (Chapter 19), other picornaviruses that infect humans have been recently discovered or previously considered to be enteroviruses and reclassified as a separate genus (Table 17.1). These genera are genetically distinct from the EV genus (Fig. 17.4) but share some physical and structural similarity with EV. On the basis of a very low genetic relationship, differences in viral proteins and processing, and a novel 2A protease, echoviruses 22 and 23 were reclassified as a new genus, Parechovirus. Additional members of this genus exist, including additional serotypes of human parechovirus,192 as well as a separate species first isolated in Swedish bank voles, Ljungan virus; Ljungan virus has been associated with diabetes in its natural host and may have a possible role in human disease.302 The human parechoviruses (HPeVs) cause a similar spectrum of illnesses as the EVs and can often be detected in cerebrospinal fluid (CSF) from meningitis cases at a frequency similar to that of the EVs.409,422 Serologic studies suggest that HPeV infection occurs at an early age, as most children were seropositive by the age of 2 years.3,198,385 HPeV3 has been associated with sepsis-like illness and CNS disease in infants.32,41
Another distinct picornavirus genus associated with human infection is Kobuvirus.434 Although little information is currently available about this virus, it appears that it is often associated with gastroenteritis in young children and infection is common. Members of the genus Cardiovirus have also been associated with disease in humans, but they do not appear to be a major cause of human illness.301,309 Viruses in the proposed genus Salivirus are related to kobuviruses and have also been associated with gastroenteritis in humans.167,250 Another proposed genus, Cosavirus, has been detected at a relatively high frequency in stool, but there is no known association with disease. What is notable about all of these newer genera is that the currently available molecular reagents for the detection of EV do not detect these viruses (see Diagnosis).
Pathogenesis and Pathology
Entry into the Host
Virus infection normally requires that the virion can attach to the cell surface, and it was long imagined that each virus would have a single receptor. For poliovirus, at least, this may be the case: the virus binds to the poliovirus receptor (PVR,279 now named CD155), a transmembrane glycoprotein in the immunoglobulin superfamily that mediates adhesion of natural killer (NK) cells, and triggers their effector functions. PVR (human CD155) appears to be the major factor regulating the virus’s natural host range, which is limited to humans and Old World primates. CD155 homologs/orthologs have been identified and characterized in mice289 and in New World monkeys; the extracellular domains share ∼70% amino acid homology with hCD155, and these homologs do not support efficient PV binding or infection. Several laboratories have generated transgenic mice that express PVR, and in many of the resulting models, PV was shown to induce neurologic disease and paralysis following parenteral administration.82,117,189,233,279,295,342,343,344,449 However, oral administration did not cause disease even when the PVR transgene was regulated by a promoter that drove protein expression in enterocytes and microfold (M) cells; PV appeared to bind to the intestinal cells, but productive infection was not observed following oral inoculation of greater than 108 plaque-forming units/mL (pfu) of virus.449 These findings are consistent with studies of humans and susceptible primates, in which hCD155 expression has been identified in many tissues, but productive infection is limited largely to the CNS. Thus, factors other than PVR expression play a key role in determining in vivo tropism.
It is now generally accepted that some viruses, or viral strains, may have more than one receptor, perhaps expanding their potential host range. EV71, most frequently associated with hand-foot-and-mouth disease in children but capable of causing devastating neurologic pathology,381 has at least two receptors: scavenger receptor B2435 and P-selectin glycoprotein ligand-1.303 Indeed, some viruses appear to interact with two different surface molecules on a single cell, perhaps in series, with one protein acting as the binding moiety, before “handing over” the virus to a second protein that facilitates its entry into the cell. This is thought to occur for some CVBs that bind to decay accelerating factor (DAF, CD55) but then must interact with another protein, the coxsackievirus and adenovirus receptor (CAR), in order to enter the cell.
Site of Primary Replication
Human enteroviruses are spread by the fecal–oral route and respiratory droplets, so systemic infection requires the virus to
cross the gastrointestinal wall, most of which is lined with epithelial cells that form a barrier to invasion. Perhaps surprisingly, given the many years of study, the primary site of PV infection and replication in the intestine remain unknown. PV has been identified in lymphoid tissues, such as the tonsils,36 and in lymphoid aggregates, commonly termed Peyer’s patches (PPs), that are present in the ileum of the small intestine. PPs are overlaid with a specialized follicle-associated epithelium (FAE) that contains M cells, which can transport certain molecules from the gastrointestinal lumen, across the epithelial layer, and into cells in the PP. Some studies suggest that PV may replicate within these epithelial cells and lymphoid cells,204 while others suggest that the virus may be transcytosed through the M cell, subsequently establishing infection in an unidentified cell in the PP.375 The cells in which CVB initially replicates are also uncertain; this issue is further clouded by the predominant use, in mouse models, of the intraperitoneal route of infection. Human rhinovirus (HRV) infects epithelial cells of the airways. Infection of the nasal epithelium causes few detectable pathologic changes, even if rhinitis is quite severe, and—as is true for many virus-induced diseases—many of the symptoms appear to be caused by the host response rather than by direct virus-mediated tissue damage.420,421
cross the gastrointestinal wall, most of which is lined with epithelial cells that form a barrier to invasion. Perhaps surprisingly, given the many years of study, the primary site of PV infection and replication in the intestine remain unknown. PV has been identified in lymphoid tissues, such as the tonsils,36 and in lymphoid aggregates, commonly termed Peyer’s patches (PPs), that are present in the ileum of the small intestine. PPs are overlaid with a specialized follicle-associated epithelium (FAE) that contains M cells, which can transport certain molecules from the gastrointestinal lumen, across the epithelial layer, and into cells in the PP. Some studies suggest that PV may replicate within these epithelial cells and lymphoid cells,204 while others suggest that the virus may be transcytosed through the M cell, subsequently establishing infection in an unidentified cell in the PP.375 The cells in which CVB initially replicates are also uncertain; this issue is further clouded by the predominant use, in mouse models, of the intraperitoneal route of infection. Human rhinovirus (HRV) infects epithelial cells of the airways. Infection of the nasal epithelium causes few detectable pathologic changes, even if rhinitis is quite severe, and—as is true for many virus-induced diseases—many of the symptoms appear to be caused by the host response rather than by direct virus-mediated tissue damage.420,421
Spread in the Host
Following replication in the alimentary tract, PV enters the blood, thereby potentially gaining access to all tissues; however, in normal hosts, viral replication is highly restricted, being readily detected mainly in the CNS.36 PV can enter the CNS in two ways: first, from the blood—the virus is thought to be able to cross the blood brain barrier (BBB), perhaps independently of the PVR,437 thereby accessing the CNS parenchyma; and second, by retrograde axonal transport, in which the virus (apparently in the form of an intact virion) ascends the neuronal axon, perhaps in endosomes, and uncoating begins when PV reaches the cell body.320,321,322 This may underpin provocation poliomyelitis, a phenomenon in which a traumatized limb is more susceptible to paralytic polio. The trauma may be directly associated with the virus, as reported in 1935, when it was noted that paralysis first appeared in (or was most severe in) the limb that had received an intramuscular inoculation of “pre-Salk” polio vaccine.244 Such inoculation poliomyelitis also was observed in the “Cutter incident,” when an incompletely inactivated Salk vaccine was administered.297 However, provocation poliomyelitis does not require that trauma and virus be administered to the same limb; when PV was administered intravascularly to monkeys that had received innocuous injections into one limb, paralysis was more likely to develop in the injected muscles.38 Provocation poliomyelitis also has been reproduced in a mouse model.139 Mechanisms other than retrograde axonal transport may contribute. For example, the peripheral trauma may increase the permeability of the BBB locally, in the region that innervates the injured muscle; this could explain why provocation poliomyelitis still occurs in traumatized limbs despite scission of the ipsilateral peroneal muscle.296 Dissemination of CVB and other enteroviruses appears to occur largely by the hematogenous route. Viremia is frequently found, when sought; this is true even in rhinovirus infection of normal or asthmatic children,432 in which spread to the lower respiratory tract was thought to occur by more direct means.
Cell and Tissue Tropism
It has been proposed that the observed in vivo tropism of PV for the CNS may result from differing efficiencies of viral internal ribosome entry site (IRES) utilization by the various cell types. However, this explanation appears to be incorrect; the PV IRES is equally functional in many cell types in vivo, including those that do not support virus replication in the living animal.211 Rather, the explanation may lie in the ability of an infected cell (or tissue) to respond to type I interferons (T1IFNs). Following PV infection, PVR transgenic mice lacking the receptor for these key antiviral cytokines developed severe lesions not only in the CNS but also in the liver, spleen, and pancreas.190 Thus, in immunocompetent PVR transgenic mice (and, by extension, in the natural hosts), the absence of apparent PV infection in most hCD155+ peripheral tissues may result from these cells’ being able to mount rapid and strong T1IFN responses; and the apparent neurotropism of the virus in immunocompetent hosts may reflect a reduced or delayed T1IFN response within the CNS.190
CVB3 can cause pancreatitis, myocarditis, and meningoencephalitis. Enteroviruses, and especially CVB, have been implicated in up to one-third of human pancreatitis cases.22,326,404 In the mouse model, the pancreas appears to be the first major site of abundant CVB3 replication, with virus titers reaching ∼104 pfu/gram after only 12 hours, and ∼1010 pfu/gram at 24 hours postinfection.277,278 As shown in Figure 17.5A, there is a large inflammatory infiltrate and widespread destruction of the exocrine pancreas, but the islets of Langerhans remain apparently unaffected. Transmission electron microscopy (Fig. 17.5B and C) reveals, in infected acinar cells, nuclear pyknosis and the accumulation of small double-membraned autophagy-like vesicles, also termed compound membrane vesicles, which are characteristic of enterovirus-infected cells. CVB4 also causes pancreatitis,339,340,406 the severity of which depends on the virus isolate that is used; a single amino acid change in VP1 appears to be largely responsible for switching the phenotype from mild pancreatitis to a more severe form.51,142 CVB has been found in acinar cells but not in islets of Langerhans356,407,410 by immunohistochemistry23,406 and by in situ hybridization.23,407,410 CAR expression correlates with the observed pathology: the receptor messenger RNA (mRNA) is expressed at very high levels in acinar cells but not in the pancreatic ducts or the islets of Langerhans (204), consistent with the observation that CVB-infected mice do not develop hyperglycemia.277,338 Cre recombinase-mediated deletion of CAR from the pancreas confers substantial, albeit incomplete, protection against organ damage during CVB infection.203
A role for EV-B, and in particular CVB, also has been suggested in type 1 diabetes (T1D), and evidence is available to support this notion.40,75,341 Several studies have implicated antibody or T-cell cross-reactivity between CVB and host proteins to explain the observed correlations.107,179,180,299,300,353 The innate immune system, too, may be involved; a human genome-wide scan for nonsynonymous single nucleotide polymorphisms, comparing healthy individuals to diabetic patients, identified MDA5 as a T1D susceptibility locus.379 However, despite many years of study, a causal role for CVB (or any enterovirus) in T1D has not been convincingly demonstrated in humans, and the issue remains controversial. It is possible that, as has been proposed for pancreatitis, enteroviruses are a co-factor in T1D, very rarely initiating disease in healthy islets but tipping
the balance in individuals who—unbeknownst to them—have ongoing islet inflammation and are in prediabetic status.
the balance in individuals who—unbeknownst to them—have ongoing islet inflammation and are in prediabetic status.
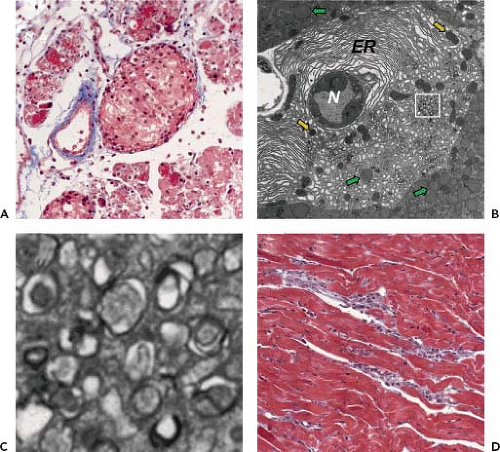 Figure 17.5. Histopathology and electron microscopy of coxsackievirus B3 (CVB3)-infected mice. A: Inflammatory infiltrate of pancreas with destruction of the exocrine pancreas but sparing of the islets of Langerhans. B: Transmission electron microscopy of infected acinar cells showing nuclear pyknosis. C: Further magnification of B showing accumulation of small double-membraned autophagy-like vesicles. D: Infected heart myocardium showing inflammatory infiltrate that contains predominantly macrophages, T cells, and natural killer cells. |
CVB has long been considered one of the principal causes of viral myocarditis,81 and this view has been confirmed in several recent reports.16,106,262,264,266 The inflammatory infiltrate (Fig. 17.5D) contains predominantly macrophages, T cells, and natural killer cells. Several mechanisms have been proposed to explain CVB-mediated cardiomyocyte destruction. The first is direct, virus-mediated damage. Cardiomyocytes can be infected in vitro,152,206 and infected cells are rapidly lysed.158 These in vitro findings are corroborated by in vivo ultrastructural studies of myocardial tissue, which show clear evidence of virus infection of cardiac muscle cells and cell death.131,163,228 The second proposed mechanism is immunopathologic damage. The inflammatory infiltrate contains CD8+ T cells, natural killer cells, and macrophages,67,129,156,370 and other studies have implicated g d T cells in CVB pathogenesis.174,175,176,178 Finally, studies have implicated autoimmunity in CVB-triggered myocarditis.176,299,353,354,355 One potential means by which this could occur is via molecular mimicry (i.e., immunologic cross-reactivity between viral and heart proteins), and there is evidence that this occurs at both the antibody121,122 and T-cell176,177,181 levels. However, recent studies in mice that lack CAR expression only on cardiomyocytes casts doubt on the importance of molecular mimicry in CVB-induced myocarditis. Thus, virus replication in the heart is a prerequisite for myocardial destruction, and this is difficult to reconcile with molecular mimicry; these data do not, of course, exclude a role for autoimmunity induced by other means, for example, by virus-driven exposure of sequestered cardiac antigens.
Immune Response
Innate Immunity
The innate immune response plays a central role in regulating virus infection, as illustrated earlier by the important role played by T1IFNs in regulating PV tissue tropism and pathogenesis. RNA viruses may trigger one (or more) of at least three sensor groups:208 Toll-like receptors (TLRs), retinoic acid–inducible gene I (RIG-I)-like receptors (RLRs), and NOD-like receptors (NLRs, some of which assemble into larger structures termed inflammasomes). The interactions between enteroviruses and NLRs have not been reported, so
herein we focus on the first two sensor groups. Triggering of TLRs and RLRs alters the expression of hundreds of genes including a variety of cytokines, chemokines, and other proteins, some of which can directly counter virus infection (e.g., protein kinase regulated by RNA [PKR, discussed later] and type I interferons), while others may regulate the development of the adaptive antiviral immune response. The roles of cell surface and internal TLRs during enterovirus infections have been evaluated in the CVB model (Fig. 17.6). TLR4 on human pancreatic cells is triggered by CVB4,398 and TLR4 knockout (KO) mice infected with CVB3 show reduced virus titers and myocarditis.109 A comparison of male and female mice confirmed that TLR4 signaling was correlated with the severity of myocarditis.118 However, the administration of TLR4 stimulants such as lipopolysaccharides (LPSs) greatly increased the severity of CVB-induced myocarditis, suggesting that CVB-mediated triggering of TLR4 in vivo is likely to be submaximal.242,346 TLR3 senses double-stranded RNA (dsRNA) molecules, which are commonly produced during the replication of RNA viruses.13 One study of CVB4 infection of TLR3-deficient mice suggested that TLR3 was almost indispensable for the innate response to this virus347 and, when compared to wild-type (wt) mice, TLR3KO mice showed increased mortality and developed more severe myocarditis following CVB3 infection.298,416 Genomic screening of patients diagnosed with enteroviral myocarditis or dilated cardiomyopathy (DCM) revealed two TLR3 sequence variants, both of which showed reduced responsiveness to ligand;120 this suggests that a strong TLR3-triggered response may protect against enteroviral myocarditis. Other internal TLRs also can contribute to the control of CVB infections. For example, human cardiac inflammatory responses to CVB are reported to be dependent largely on TLR7 and TLR8,397 both of which recognize single-stranded RNA (ssRNA) and other small molecules.151 Contrary to the reported beneficial effects of a strong TLR3 response to enteroviruses, a strong TLR8 response may be associated with adverse outcomes in patients with enterovirus-associated DCM.365 Autophagy is up-regulated by TLRs, and the most potent pro-autophagic effects are mediated by ssRNA/TLR7 signaling.91 Electron microscopic (EM) studies of poliovirus-infected cells revealed an association between PV and double-membraned intracellular vesicles,88 subsequently shown to be autophagy related.368 Extensive membrane remodeling occurs in a poliovirus-infected cell, mediated by the viral 2BC and 3A proteins,383 and the resulting vesicles carry several autophagosome-related proteins.391,392 A similar relationship between CVB and autophagic vesicles has been reported,134 and there is a marked increase in double-membraned structures within CVB-infected cells, both in tissue culture424,441 and in vivo.214 Although autophagy is generally thought to support the replication of these viruses, to date, the effects have been determined only in tissue culture cells and are extremely modest; inhibiting autophagy in cells infected with PV,193 CVB3,424 CVB4,441 or EV71173 reduces production of each of these viruses by only 1.5- to 4-fold. To date, few studies have evaluated autophagy during enteroviral replication in vivo.
herein we focus on the first two sensor groups. Triggering of TLRs and RLRs alters the expression of hundreds of genes including a variety of cytokines, chemokines, and other proteins, some of which can directly counter virus infection (e.g., protein kinase regulated by RNA [PKR, discussed later] and type I interferons), while others may regulate the development of the adaptive antiviral immune response. The roles of cell surface and internal TLRs during enterovirus infections have been evaluated in the CVB model (Fig. 17.6). TLR4 on human pancreatic cells is triggered by CVB4,398 and TLR4 knockout (KO) mice infected with CVB3 show reduced virus titers and myocarditis.109 A comparison of male and female mice confirmed that TLR4 signaling was correlated with the severity of myocarditis.118 However, the administration of TLR4 stimulants such as lipopolysaccharides (LPSs) greatly increased the severity of CVB-induced myocarditis, suggesting that CVB-mediated triggering of TLR4 in vivo is likely to be submaximal.242,346 TLR3 senses double-stranded RNA (dsRNA) molecules, which are commonly produced during the replication of RNA viruses.13 One study of CVB4 infection of TLR3-deficient mice suggested that TLR3 was almost indispensable for the innate response to this virus347 and, when compared to wild-type (wt) mice, TLR3KO mice showed increased mortality and developed more severe myocarditis following CVB3 infection.298,416 Genomic screening of patients diagnosed with enteroviral myocarditis or dilated cardiomyopathy (DCM) revealed two TLR3 sequence variants, both of which showed reduced responsiveness to ligand;120 this suggests that a strong TLR3-triggered response may protect against enteroviral myocarditis. Other internal TLRs also can contribute to the control of CVB infections. For example, human cardiac inflammatory responses to CVB are reported to be dependent largely on TLR7 and TLR8,397 both of which recognize single-stranded RNA (ssRNA) and other small molecules.151 Contrary to the reported beneficial effects of a strong TLR3 response to enteroviruses, a strong TLR8 response may be associated with adverse outcomes in patients with enterovirus-associated DCM.365 Autophagy is up-regulated by TLRs, and the most potent pro-autophagic effects are mediated by ssRNA/TLR7 signaling.91 Electron microscopic (EM) studies of poliovirus-infected cells revealed an association between PV and double-membraned intracellular vesicles,88 subsequently shown to be autophagy related.368 Extensive membrane remodeling occurs in a poliovirus-infected cell, mediated by the viral 2BC and 3A proteins,383 and the resulting vesicles carry several autophagosome-related proteins.391,392 A similar relationship between CVB and autophagic vesicles has been reported,134 and there is a marked increase in double-membraned structures within CVB-infected cells, both in tissue culture424,441 and in vivo.214 Although autophagy is generally thought to support the replication of these viruses, to date, the effects have been determined only in tissue culture cells and are extremely modest; inhibiting autophagy in cells infected with PV,193 CVB3,424 CVB4,441 or EV71173 reduces production of each of these viruses by only 1.5- to 4-fold. To date, few studies have evaluated autophagy during enteroviral replication in vivo.
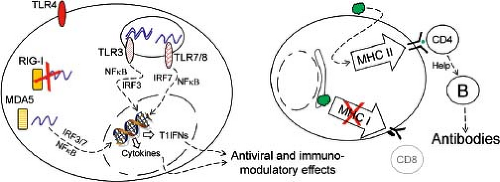 Figure 17.6. Signaling through Toll-like receptors (TLRs) during enterovirus infections. TLR4 on human pancreatic cells is triggered by infection. TLR3 senses double-stranded RNA (dsRNA) molecules and a strong TLR3-triggered response may protect against enteroviral myocarditis. Other internal TLRs also can contribute to the control of coxsackievirus B (CVB) infections. |
Three RLRs have been identified: RIG-I, MDA5 (melanoma differentiation–associated gene 5), and LGP2 (laboratory of genetics and physiology 2). Unlike TLRs, these proteins are expressed in most cell types. All are activated by nucleic acids, and all are cytosolic, although it has been reported that RIG-I co-localizes with F-actin, and thus is associated with the cytoskeleton.293 Many RNA viruses produce abundant dsRNA and ssRNA with 5′ triphosphate and, therefore, strongly activate RIG-I. In the absence of RIG-I, the innate response to several RNA virus families is abrogated.257 The 5′ terminus of an enteroviral RNA lacks the 5′ triphosphate moiety, instead bearing a modified protein (VPg), and, for this reason, these viruses do not stimulate RIG-I. Infected cells appear to rely largely on MDA5 to alert them to the presence of picornaviral RNA. This sensor is tripped by the cardiovirus encephalomyocarditis virus (EMCV),127,210 and two recent publications using MDA5 knockout mice suggest that enteroviruses, too, trigger MDA5 signaling.183,414 MDA5, like RIG-I, is degraded in poliovirus-infected HeLa cells, providing one possible mechanism by which enteroviruses might paralyze the innate immune response.28 However, the observation that MDA5 deficiency has a marked effect during CVB3 infection suggests that, if MDA5 degradation occurs in CVB-infected cells, the process does not prevent MDA5-mediated triggering of the innate response.
At least two observations have been made that may be relevant to both TLR- and RLR-mediated responses to CVB (and, possibly, other enteroviruses). First, many cells—exemplified by macrophages and dendritic cells—are phagocytic and engulf dead or dying cells. Thus, although human dendritic cells (DCs) cannot be productively infected by CVB,239 these cells can consume debris from CVB-infected cells, thereby potentially introducing viral materials to the cytosolic and intravesicular sensors, potentially inducing an antiviral state in the DCs.238 Second, as well as degrading the sensors themselves, some enteroviruses can interrupt the downstream signaling upon which the innate response depends. Signaling via TLR3 and the RLRs rely, respectively, on proteins named TRIF and MAVS, both of which are degraded in CVB-infected cells, apparently by the viral 3C protease.292
NK cells, which are part of the innate response to many infections, are important in protecting against CVB-induced pancreatitis in the mouse model.408 The importance of NK cells in combating human enteroviral infections is unknown, but human NK cells can produce interferon-g (IFN-g) in response to CVB-infected cells.182
Adaptive Immunity
Antibodies and CD8+ T cells together provide a strong antigen-specific barrier against virus infections. Under normal circumstances, these two arms of the adaptive response complement each other. However, members of the Enterovirus species appear to be an exception to this general rule. Patients with X-linked agammaglobulinemia are highly susceptible to enteroviral infections,282 and after receiving live poliovirus vaccine, such individuals may continue to shed virulent poliovirus for up to ∼20 years.219,267 The near-absolute requirement for antibodies in protecting against enteroviruses has been confirmed in an animal model of CVB3 infection, using B-cell knockout (BcKO) mice; these mice cannot eradicate the virus, and high titers are present in many organs.278 These observations suggest that, for agammaglobulinemic hosts infected with an enterovirus, there may be some deficit in the backup system that, for most viruses, is provided by CD8+ T-cell responses. This may, in part, explain why most studies of enteroviral infections, in mouse and man, have identified strong antibody responses, while CD8+ T-cell responses—so easily detected in most virus infections—are weak (if detected at all). The enterovirus genus is large, and the adaptive responses to only a select few enteroviral species will be discussed later.
Human Enterovirus A
The best-studied pathogen in this species is EV71. The virus triggers an immunoglobulin M (IgM) response that is detectable as early as 2 days postinfection,399 as well as a strong neutralizing IgG response that recognizes epitopes in the N-terminal segment of VP1.387 Neutralizing antibodies, when transferred to uninfected neonatal recipient mice, are able to protect against a lethal challenge infection.445 Studies of knockout mice showed that B cells, in particular, were important for survival following EV71 infection, and B-cell–deficient mice treated with virus-specific antibody either before or during EV71 infection had lower virus titers, less severe disease, and lower mortality.253 Memory T-helper 1 (Th1) CD4+ T-cell responses specific for three epitopes in VP1 have been identified in EV71-positive individuals,116 but no CD8+ T-cell epitopes have been reported.
Human Enterovirus B
CVBs are the best-studied pathogens in this species and, as described earlier, can trigger severe acute and chronic diseases including myocarditis, DCM, pancreatitis, and aseptic meningitis. Infection by these viruses triggers a rapid and strong neutralizing antibody response. Virus-specific IgM appears during the first week of infection, followed by a strong neutralizing IgG response. The CVB3-specific IgM titer wanes over time, but IgG antibodies persist.256 Work in T-cell–deficient (nude) mice indicates that at least some of the CVB-specific antibody response is T-cell independent,150,364,426 although some studies suggest that CD4+ T-cell help may be important for the induction of strong neutralizing antibody responses.246 B cells appear to be targeted by CVB278 and may provide a reservoir for the virus during persistent CVB infection. Viral RNA-positive cells, most probably B cells, can be found in the splenic follicles and germinal centers;14,195,207,229,278 approximately 1% of B cells are infected with CVB3 in vivo, and these cells may accelerate the systemic distribution of virus.278 T cells can help control CVB infection, although much less effectively than antibodies. In vivo analyses of CVB-specific T cells has been challenging because these viruses, despite replicating to high titers in mice, induce remarkably weak CD8+ T-cell responses.378,213a Nevertheless, some responses can be identified, exemplified by CD4+ T-cell responses against epitopes expressed by CVB4.143,144 CD8+ T-cell responses are particularly meager. Epitopes in several viral proteins have been identified in human studies, but their detection required ∼2 weeks of in vitro peptide antigen restimulation.416
Human Enterovirus C
This species contains a number of coxsackie A viruses, but the most important pathogen is PV, which induces a strong neutralizing antibody response that is necessary to control the infection. Virus infection and vaccination induce strong and long-lasting humoral responses,79 but the immunity is not sterilizing and secondary infections of the gut can occur. Susceptibility to such reinfections, and subsequent shedding of PV, may be controlled by IgA.48 PV-driven T-cell proliferation has been observed in infant vaccinees, and the responses appear to be cross-reactive across different enteroviruses.202 However, PV-specific T-cell responses in OPV-vaccinated infants may be weaker than those in adults.405 OPV induces major histocompatibility complex (MHC) class II–restricted memory CD4+ T-cell responses targeted to epitopes in all four capsid proteins.377 CD8+ T-cell responses to PV vaccination are long-lived but, as is true for most CVB-specific responses, they became detectable only after several rounds of in vitro restimulation, suggesting that T-cell numbers in vivo were low.413 Mice are not naturally infected by PV, but PVR-transgenic (PVR-Tg) animals have allowed the analysis of T- and B-cell responses and their roles in antiviral protection. Adoptive transfer of PV-primed B cells together with a VP4-specific CD4+ T-cell clone protected PVR-Tg mice against a lethal challenge of PV, but neither cell population alone was protective, indicating that the virus-specific B cells required T-cell help.265
Release from Host
Virion assembly and RNA packaging of enteroviruses remain very poorly understood. Although enteroviruses are generally considered to be highly lytic—and, therefore, released from cells upon their lysis—there is some evidence from cell culture studies consistent with PV release by nonlytic means.330,401 It has been proposed that autophagy-mediated release of PV may occur, permitting the virus to exit a cell in a noncytolytic manner;193 this proposed mechanism has, memorably, been termed AWOL (autophagy-mediated exit without lysis).390
Virulence
Viral virulence is a complex interplay between virus and host, and some of the contributing elements, such as receptor distribution and host responses, have been discussed earlier. Here, we will focus on selected enteroviral sequences and proteins and their contributions to a virulent phenotype. Wild-type PV is more neurovirulent than the attenuated viruses that constitute the oral vaccine, and studies have identified, in all three PV serotypes, changes in the 5′ noncoding region that can alter neurovirulence. For example, sequence comparison between the attenuated Sabin 3 virus and revertants from cases of
vaccine-associated poliomyelitis showed that a single U-C change in the viral IRES, at position 472, conferred a growth advantage in the human intestine and resulted in increased neurovirulence, although, on its own, the change was insufficient to confer full neurovirulence;108 a second change, leading to an amino acid substitution in VP3, almost completely restored virulence.417 Mutations in the IRES of PV types 1 and 2 also modulate the neurovirulent phenotype.212,260,261 The in vivo neuroattenuating phenotype imposed by changes in the IRES, together with tissue culture studies showing apparent cell-specific effects of the IRES mutations,146 led to the proposal that neuroattenuation might be explained by a neuron-specific reduction in usage of the mutated IRES. However, in vivo analyses have demonstrated that, while the Sabin 3 IRES sequence is indeed less effectively utilized in neurons, this defect also is observed in other cells and tissues.212 Thus, another explanation was sought. The importance of T1IFNs in modulating PV neurovirulence in vivo has been described earlier. Cardiovirulence in CVB3 also has been mapped to various locations in the 5′ nontranslated region (NTR), ∼80 to 240 bases from the 5′ end of the genome,64,104,400 although the capsid region, too, plays a part.395 In contrast to the extensive mapping of the limits of, and functional domains within, the PV IRES, analysis of the CVB IRES has been limited.
vaccine-associated poliomyelitis showed that a single U-C change in the viral IRES, at position 472, conferred a growth advantage in the human intestine and resulted in increased neurovirulence, although, on its own, the change was insufficient to confer full neurovirulence;108 a second change, leading to an amino acid substitution in VP3, almost completely restored virulence.417 Mutations in the IRES of PV types 1 and 2 also modulate the neurovirulent phenotype.212,260,261 The in vivo neuroattenuating phenotype imposed by changes in the IRES, together with tissue culture studies showing apparent cell-specific effects of the IRES mutations,146 led to the proposal that neuroattenuation might be explained by a neuron-specific reduction in usage of the mutated IRES. However, in vivo analyses have demonstrated that, while the Sabin 3 IRES sequence is indeed less effectively utilized in neurons, this defect also is observed in other cells and tissues.212 Thus, another explanation was sought. The importance of T1IFNs in modulating PV neurovirulence in vivo has been described earlier. Cardiovirulence in CVB3 also has been mapped to various locations in the 5′ nontranslated region (NTR), ∼80 to 240 bases from the 5′ end of the genome,64,104,400 although the capsid region, too, plays a part.395 In contrast to the extensive mapping of the limits of, and functional domains within, the PV IRES, analysis of the CVB IRES has been limited.
Persistence
Although both PV and CVB are rapidly cytolytic in many of the cell types that they infect, both viruses can establish persistent or chronic infection in tissue culture.78,119,269,331 In neuroblastoma cells, PV persistence is associated with accrual of mutations in the capsid region,330 and for both PV and CVB, moving from a cytolytic to persistent phenotype in cell culture may be inversely related to the capacity of the virus to adsorb to the receptor and/or to the level of receptor expression.53,240 It recently has been reported that, during persistent infection of cultured cells that express low levels of CAR, CVB accumulated changes in the capsid that allow the variant virus to bind to novel (non-DAF, non-CAR) molecules, and this more promiscuous activity conferred a replicative advantage upon the variant.52 Cellular factors contribute to the establishment of PV persistence.126 Cell cycle status may play a role in the case of CVB, which does not replicate efficiently in tissue culture cells rendered quiescent by drugs or by serum starvation but undergoes productive and cytolytic replication when the cell cycle is triggered.112,113
Enteroviral persistent infections can take place under two general scenarios. First, a chronic productive enterovirus infection can occur in immunocompromised hosts. As noted earlier, agammaglobulinemic individuals who receive oral polio vaccine may retain, and excrete, the virus for many years. One study of individuals with primary immunodeficiency who had developed vaccine-associated paralytic poliomyelitis found that approximately one in five secreted vaccine-derived PV at 6 months after their last OPV dose, but this frequency declined to 0% when the interval was 10 years.222 The low prevalence of the underlying condition means that such persons are rare; only ∼40 such individuals have been identified.429 Fortunately, a more common potential cause of immunosuppression, human immunodeficiency virus (HIV) infection, seems not to correlate with PV persistence/shedding.24,157 Second, immunocompetent hosts may carry virus (or at least viral RNA) for many years. Given the frequency of enteroviral infections, it is reasonable to suppose that this is the more common of the two types of enterovirus persistence. In the vast majority of cases in which enteroviral persistence in vivo has been reported in immunocompetent hosts, infectious virus was not identified; rather, viral materials (most commonly RNA) were reported and infectious particles, if sought, were not found. CVB RNA has frequently been detected by PCR in many analyses of cardiac biopsies from individuals with Dcm20,21,45,232 or inflammatory peripheral myopathy.44 From results obtained in a murine model of polymyositis, the authors concluded that the RNA was maintained in double-stranded form, with little indication of virus mutation/evolution.386 However, recent analyses of CVB3 genomes isolated from the hearts of persistently infected mice suggest that CVB persistence in vivo may be dependent upon the deletion of nucleotides at the 5′ end of the genome.226 Several deletions were reported, some extending to nucleotide 49, and all affecting the 5′ cloverleaf structure that is considered vital for RNA replication. Importantly, the materials were infectious; although replicating very slowly, they could be maintained in culture and did not require a helper virus. The VPg protein was present on several of the deletion mutant genomes and, notably, ∼25% of RNA encapsidated into virions was negative sense. The authors proposed that the encapsidation of negative strands might occur because the terminal deletions, by altering RNA replication, markedly reduced the ratio of positive to negative strands. Similar 5′ terminal deletion variants subsequently were identified in CVB3 that had been passed in primary tissue culture cells225 and, critically, in a CVB2 genome isolated from the heart of a human who had succumbed to enteroviral myocarditis.63 The poor infectivity of these mutated viruses may explain why infectious virus was not identified in the vast majority of previous studies in which enterovirus RNA was found. To date, there is no evidence to suggest that these terminally deleted variants can be transmitted under normal circumstances.
Epidemiology
Demographics
Despite the nearly ubiquitous nature of EV infections and the wide variety of clinical presentations, the demographics of the various infections and diseases have some consistent characteristics. In particular, several factors, including age, sex, and socioeconomic status, have largely predictable effects.
One of the most important determinants of EV infection outcome is age. Different age groups have different susceptibilities to infection, severity of illness, clinical manifestations, and prognoses following EV infection. Understanding these age effects on outcome of infection is complicated by the widely divergent prior history of infection and resulting immunity. Nevertheless, it is possible to make certain generalizations.
The largest amount and duration of virus shedding occurs on primary infection with a given EV serotype. Because infection is so common, most primary infections occur during childhood. For these reasons, young children are probably the most important transmitters of EV, particularly within households. The greater exposure of children to virus during infection may make them more likely to have significant clinical
symptoms. For example, in outbreaks of meningitis, children typically have higher rates of disease than adults.134,191 Most studies, however, do not separately determine age-specific infection rates and disease rates, and the relative rates at which adults are infected are not generally known.
symptoms. For example, in outbreaks of meningitis, children typically have higher rates of disease than adults.134,191 Most studies, however, do not separately determine age-specific infection rates and disease rates, and the relative rates at which adults are infected are not generally known.
The incidence of poliomyelitis is relatively low for the first 4 to 6 months of life in countries in which control through vaccination has not yet been achieved, because of the frequent presence of protective maternal antibody. In these countries, an increased incidence is seen of paralytic disease in children older than 6 months compared with children in wealthier developed countries, presumably related to an earlier exposure to virus as a result of poor sanitary conditions. Ironically, areas with improved hygiene may have a decrease in infant exposure, leaving an older (unexposed) population susceptible to epidemic disease, with high rates of paralytic disease during an outbreak.335 Adults are more likely to be severely affected in both developing and developed countries, tending to acquire paralytic poliomyelitis rather than nonparalytic CNS disease (i.e., aseptic meningitis), abortive illness, or asymptomatic infection.56,57,170 The reason for the increase in severity later in life is unknown. A possible reason relates to the finding that fast axonal flow, which appears important in the spread of PV within the CNS,200 increases with age. In addition, it may be that CD155 expression or host factors important in replication change with age.
Severity of a number of enteroviral diseases besides poliomyelitis may be strikingly age related. An indirect indication is that a delay in first infection with a number of EVs increases risk of more severe disease. For example, exanthema associated with CVA and echoviruses are for the most part milder in children than in adults. On the other hand, some EVs cause more severe disease in newborns than in older children and adults, possibly inducing a fulminant viral sepsis with myocarditis, encephalitis, and sometimes death (see Clinical Features: Neonate and Infant Disease).68,194 In addition, recent outbreaks of hand-foot-and-mouth disease caused by EV71 have been associated with a significant CNS complication, fatal brainstem encephalitis, that was restricted largely to young children (Table 17.3) (see Clinical Features: Meningitis and Encephalitis).235,258,428
In general, encephalitis and aseptic meningitis caused by EV appear to be most frequent among those 5 to 14 years of age rather than those older or younger. In a 10-year surveillance summary from the United States,286 adults tended to be overrepresented among cases of severe disease (paralysis, encephalitis, meningitis, carditis) when compared with the age distribution of the EV-infected population as a whole. In another study, the mean age among patients with CVB meningitis (7.7 years) or pericarditis (9.9 years) was greater than the mean age of patients with CVB gastroenteritis (1.3 years).96
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


