Endocrine system
The endocrine system consists of glands, specialized cell clusters, hormones, and target tissues. The glands and cell clusters secrete hormones and chemical transmitters in response to stimulation from the nervous system and other sites. Together with the nervous system, the endocrine system regulates and integrates the body’s metabolic activities and maintains internal homeostasis. Each target tissue has receptors for specific hormones. Hormones connect with the receptors, and the resulting hormone-receptor complex triggers the target cell’s response.
Hormonal regulation
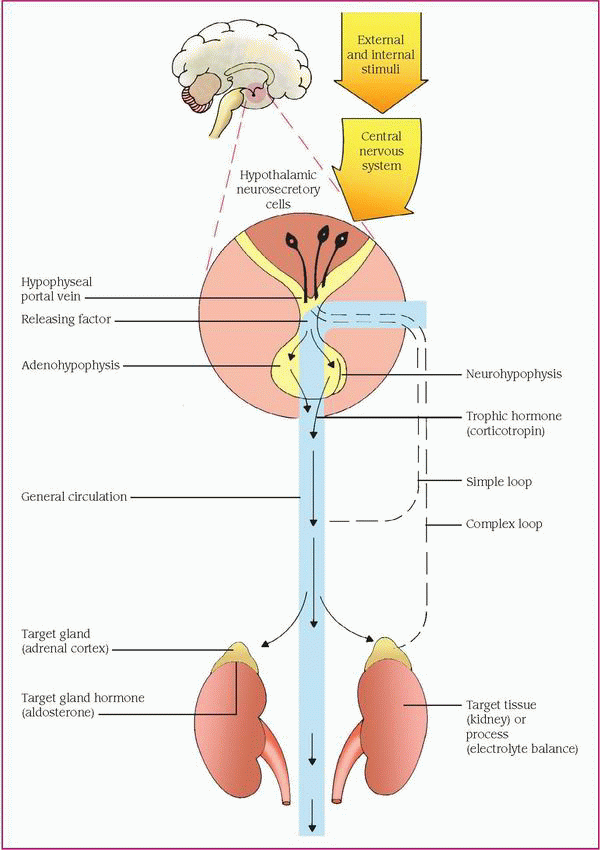
The hypothalamus, the main integrative center for the endocrine and autonomic nervous systems, helps control some endocrine glands by neural and hormonal pathways. Neural pathways connect the hypothalamus to the posterior pituitary gland, or neurohypophysis. Neural stimulation of the posterior pituitary causes the secretion of two effector hormones: antidiuretic hormone (ADH, also known as vasopressin) and oxytocin.
The hypothalamus also exerts hormonal control at the anterior pituitary gland, or adenohypophysis, by releasing and inhibiting hormones and factors, which arrive by a portal system. Hypothalamic hormones stimulate the pituitary gland to synthesize and release trophic hormones, such as corticotropin (also called adrenocorticotropic hormone), thyroid-stimulating hormone (TSH), and gonadotropins, such as luteinizing hormone (LH) and follicle-stimulating hormone (FSH). Secretion of trophic hormones stimulates the adrenal cortex, thyroid gland, and gonads. Hypothalamic hormones also stimulate the pituitary gland to release or inhibit the release of effector hormones, such as growth hormone (GH) and prolactin.
In a patient with a possible endocrine disorder, this complex hormonal sequence requires careful assessment to identify the dysfunction, which may result from defects in the gland; defects of releasing, trophic, or effector hormones; or defects of the target tissue. Hyperthyroidism, for example, may result from excessive thyrotropinreleasing hormone, TSH, or thyroid hormones, or excessive response of the thyroid gland.
Besides hormonal and neural controls, a negative feedback system regulates the endocrine system. (See Feedback mechanism of the endocrine system, page 464.)
The feedback mechanism may be simple or complex. Simple feedback occurs when the level of one substance regulates secretion of a hormone. For example, a low serum calcium level stimulates the parathyroid glands to secrete parathyroid hormone (PTH), and a high serum calcium level inhibits PTH secretion.
One example of complex feedback occurs through the hypothalamic-pituitary target organ axis. Secretion of the hypothalamic corticotropin- releasing hormone releases pituitary corticotropin, which in turn stimulates adrenal cortisol secretion. Subsequently, an increase in the serum cortisol level inhibits corticotropin by decreasing corticotropin-releasing hormone secretion or corticotropin directly. Corticosteroid therapy disrupts the hypothalamic-pituitary-adrenal
axis by suppressing the hypothalamic-pituitary secretion mechanism. Because abrupt withdrawal of steroid therapy doesn’t allow time for recovery of the hypothalamicpituitary-adrenal axis to stimulate cortisol secretion, it can induce life-threatening adrenal crisis.
axis by suppressing the hypothalamic-pituitary secretion mechanism. Because abrupt withdrawal of steroid therapy doesn’t allow time for recovery of the hypothalamicpituitary-adrenal axis to stimulate cortisol secretion, it can induce life-threatening adrenal crisis.
RHYTHMS
The endocrine system is also controlled by rhythms, many of which last 24 hours (circadian). Circadian rhythm control of corticotropin and cortisol increases levels of these hormones in the early morning hours and decreases them in the late afternoon. Stress, caused by pyrogens, surgery, hypoglycemia, exercise, severe emotional trauma, and other conditions, enhances corticotropin release and abolishes corticotropin circadian rhythmicity. High-dose glucocorticoid administration suppresses stress-related corticotropin release. An infradian rhythm is a biorhythm that repeats in patterns that exceed a 24-hour period. The menstrual cycle is an example of an infradian rhythm—in this case, 28 days.
HORMONAL EFFECTS
The posterior pituitary gland secretes oxytocin and ADH. Oxytocin stimulates contraction of the uterus and causes the milk-letdown reflex in breast-feeding women. ADH controls the concentration of body fluids by altering the permeability of the distal and collecting tubules of the kidneys to conserve water. ADH secretion depends on plasma osmolality, the characteristic of a solution determined by the ionic concentration of the dissolved substance and the solution, which is monitored by hypothalamic neurons. Hypovolemia and hypotension are the most powerful stimulators of ADH release. Other stimulators include trauma, nausea, morphine, tranquilizers, certain anesthetics, positivepressure breathing, pain, and stress.
In addition to the trophic hormones, the anterior pituitary gland secretes prolactin, which stimulates milk secretion, and GH. GH affects most body tissues. It triggers growth by stimulating protein synthesis and fat mobilization, and by decreasing carbohydrate use by muscle and fat tissue. The thyroid gland synthesizes and secretes the iodinated hormones, thyroxine and triiodothyronine. Thyroid hormones are necessary for normal growth and development, and act on many tissues to increase metabolic activity and protein synthesis.
The parathyroid glands secrete PTH, which regulates calcium and phosphate metabolism. PTH elevates the serum calcium level by stimulating resorption of calcium and excretion of phosphate from bone, and enhances absorption of calcium from the GI tract by stimulating the conversion of vitamin D to its most active form. Calcitonin, another hormone that the thyroid gland secretes, affects calcium metabolism, although its precise role in humans is unknown.
The pancreas produces glucagon from the alpha cells and insulin from the beta cells. Glucagon, the hormone of the fasting state, releases stored glucose from the liver to increase the blood glucose level. Insulin, the hormone of the postprandial state, facilitates glucose transport into the cells, promotes glucose storage, stimulates protein synthesis, and enhances free fatty acid uptake and storage.
The adrenal cortex secretes mineralocorticoids, glucocorticoids, and sex steroid hormones (androgens). Aldosterone, a mineralocorticoid, regulates the reabsorption of sodium and the excretion of potassium by the kidneys. Although affected by corticotropin, aldosterone is mainly regulated by the renin-angiotensin system. Together, aldosterone, angiotensin II, and renin may be implicated in the pathogenesis of hypertension.
Cortisol, a glucocorticoid, stimulates gluconeogenesis, increases protein breakdown and free fatty acid mobilization, suppresses the immune response, and facilitates an appropriate response to stress.
The adrenal medulla is an aggregate of nervous tissue that produces the catecholamines epinephrine and norepinephrine, which cause vasoconstriction. In addition, epinephrine stimulates the fight-or-flight response—dilation of bronchioles and increased blood pressure, blood glucose level, and heart rate. The adrenal cortex as well as the gonads secrete androgens, which are steroid sex hormones. In men and premenopausal women, the contribution of adrenal androgens is small, but in postmenopausal women, the adrenals are the major source of sex hormones.
The testes synthesize and secrete testosterone in response to gonadotropic hormones, especially LH, from the anterior pituitary gland; spermatogenesis occurs in response to FSH. The ovaries produce sex steroid hormones (primarily estrogen and progesterone) in response to anterior pituitary trophic hormones.
Pathophysiologic changes
Alterations in hormone levels, either significantly high or low, may result from various causes. Feedback systems may fail to function properly or may respond to the wrong signals. Dysfunction of an endocrine gland may manifest as either
failure to produce adequate amounts of active hormone or excessive synthesis or release. After the hormones are released, they may be degraded at an altered rate or inactivated by antibodies before reaching the target cell. Abnormal target cell responses include receptor-associated alterations and intracellular alterations.
failure to produce adequate amounts of active hormone or excessive synthesis or release. After the hormones are released, they may be degraded at an altered rate or inactivated by antibodies before reaching the target cell. Abnormal target cell responses include receptor-associated alterations and intracellular alterations.
RECEPTOR-ASSOCIATED ALTERATIONS
Receptor-associated alterations have been associated with water-soluble hormones (peptides) and involve:
♦ fewer receptors, resulting in diminished or defective hormone-receptor binding
♦ impaired receptor function, resulting in insensitivity to the hormone
♦ presence of antibodies against specific receptors, either reducing available binding sites or mimicking hormone action and suppressing or exaggerating target cell response
♦ unusual expression of receptor function.
INTRACELLULAR ALTERATIONS
Intracellular alterations involve the inadequate synthesis of the second messenger needed to convert the hormonal signal into intracellular events. The two different mechanisms that may be involved include:
♦ faulty response of target cells for water-soluble hormones to hormone-receptor binding and failure to generate the required second messenger
♦ abnormal response of the target cell to the second messenger and failure to express the usual hormonal effect.
Pathophysiologic aberrations affecting target cells for lipid-soluble (steroid) hormones are less common or may not be as easily recognized.
Disorders
Common dysfunctions of the endocrine system are classified as hypofunction and hyperfunction, inflammation, and tumor.
ADRENAL HYPOFUNCTION
Adrenal hypofunction is classified as primary or secondary. Primary adrenal hypofunction or insufficiency (Addison’s disease) originates within the adrenal gland and is characterized by the decreased secretion of mineralocorticoids, glucocorticoids, and androgens. Secondary adrenal hypofunction is caused by a disorder outside the gland, such as impaired pituitary secretion of corticotropin. It’s characterized by decreased glucocorticoid secretion. The secretion of aldosterone, the major mineralocorticoid, is commonly unaffected.
Addison’s disease is relatively uncommon and can occur at any age and in both sexes. Secondary adrenal hypofunction occurs when a patient abruptly stops long-term exogenous steroid therapy or when the pituitary is injured by a tumor or by infiltrative or autoimmune processes — these occur when circulating antibodies react specifically against adrenal tissue, causing inflammation and infiltration of the cells by lymphocytes. With early diagnosis and adequate replacement therapy, the prognosis for both primary and secondary adrenal hypofunction is good.
Adrenal crisis (addisonian crisis), a critical deficiency of mineralocorticoids and glucocorticoids, generally follows acute stress, sepsis, trauma, surgery, or the omission of steroid therapy in patients who have chronic adrenal insufficiency. Adrenal crisis is a medical emergency that needs immediate, vigorous treatment.
Autoimmune Addison’s disease is most common in white females, and a genetic predisposition is likely. It’s more common in patients with a familial predisposition to autoimmune endocrine diseases. Addison’s disease affects 1 in 16,000 neonates; it also affects 8 in 100,000 adults, with males and females affected equally.
Causes
Primary and secondary adrenal hypofunction and adrenal crisis have different causes. The most common cause of primary hypofunction is Addison’s disease that results in destruction of more than 90% of both adrenal glands, usually caused by an autoimmune process in which circulating antibodies react specifically against the adrenal tissue.
Other causes include:
♦ bilateral adrenalectomy
♦ family history of autoimmune disease (may predispose the patient to Addison’s disease and other endocrinopathies)
♦ hemorrhage into the adrenal gland
♦ infections (human immunodeficiency virus [HIV], histoplasmosis, cytomegalovirus [CMV])
♦ neoplasms
♦ tuberculosis (once the chief cause, now responsible for less than 20% of adult cases).
Causes of secondary hypofunction (glucocorticoid deficiency) include:
♦ abrupt withdrawal of long-term corticosteroid therapy (long-term exogenous corticosteroid stimulation suppresses pituitary corticotropin secretion, resulting in adrenal gland atrophy)
♦ hypopituitarism (causing decreased corticotropin secretion)
Adrenal crisis (acute adrenal insufficiency) is the most serious complication of Addison’s disease. It may occur gradually or suddenly.
Risk factors
This potentially lethal condition usually develops in patients who:
♦ don’t respond to hormone replacement therapy.
♦ undergo extreme stress without adequate glucocorticoid replacement.
♦ abruptly stop hormone therapy.
♦ undergo trauma.
♦ undergo bilateral adrenalectomy.
♦ develop adrenal gland thrombosis after a severe infection (Waterhouse-Friderichsen syndrome).
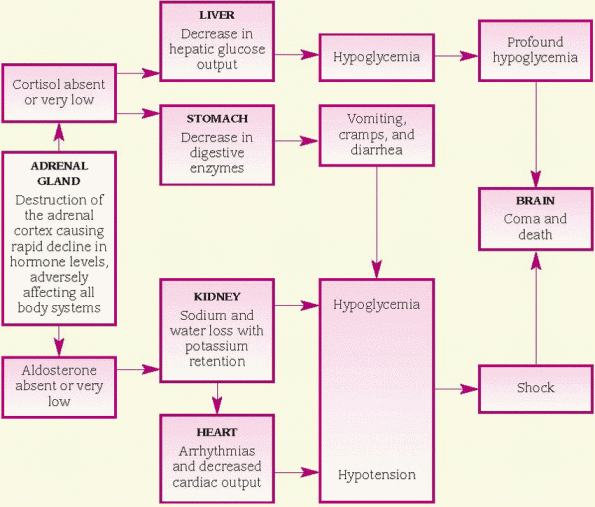
♦ exhausted body stores of glucocorticoids in a person with adrenal hypofunction after trauma, surgery, or other physiologic stress. (See Understanding adrenal crisis.)
Pathophysiology
Addison’s disease is a chronic condition that results from the partial or complete destruction of the adrenal cortex. It manifests as a clinical syndrome in which the symptoms are associated with deficient production of the adrenocortical hormones, cortisol, aldosterone, and androgens. High levels of corticotropin and corticotropinreleasing hormone accompany the low glucocorticoid level.
Corticotropin acts primarily to regulate the adrenal release of glucocorticoids (primarily cortisol); mineralocorticoids, including aldosterone; and sex steroids that supplement those that the gonads produce. Corticotropin secretion is controlled by corticotropin-releasing hormone
from the hypothalamus and by negative feedback control by the glucocorticoids.
from the hypothalamus and by negative feedback control by the glucocorticoids.
Addison’s disease involves all zones of the cortex, causing deficiencies of the adrenocortical secretions, glucocorticoids, androgens, and mineralocorticoids.
Manifestations of adrenocortical hormone deficiency become apparent when 90% of the functional cells in both glands are lost. Usually, cellular atrophy is limited to the cortex, although medullary involvement may occur, resulting in catecholamine deficiency. Cortisol deficiency causes decreased liver gluconeogenesis (the formation of glucose from molecules that aren’t carbohydrates). The resulting low blood glucose level can become dangerously low in patients who take insulin routinely.
Aldosterone deficiency causes increased renal sodium loss and enhances potassium reabsorption. Sodium excretion causes a reduction in water volume that leads to hypotension. Patients with Addison’s disease may have normal blood pressure when in a supine position but show marked hypotension and tachycardia after standing for several minutes. Low plasma volume and arteriolar pressure stimulate renin release and a resulting increased production of angiotensin II.
Androgen deficiency may decrease hair growth in axillary and pubic areas as well as on the extremities of women. The metabolic effects of testicular androgens make such hair growth less noticeable in men.
Addison’s disease is a decrease in the biosynthesis, storage, or release of adrenocortical hormones. In about 80% of the patients, an autoimmune process causes partial or complete destruction of both adrenal glands. Autoimmune antibodies can block the corticotropin receptor or bind with corticotropin, preventing it from stimulating adrenal cells. Infection is the second most common cause of Addison’s disease, specifically tuberculosis, which causes about 20% of the cases. Other conditions that can cause Addison’s disease include HIV infection, systemic fungal infections, CMV, adrenal tumor, and metastatic cancers. Infection can impair cellular function and affect corticotropin at any stage of regulation.
Signs and symptoms
Clinical features vary with the type of adrenal hypofunction.
Primary hypofunction
♦ Weakness and fatigue caused by alterations in adrenal hormone balance
♦ Weight loss, nausea, vomiting, and anorexia resulting from glucocorticoid deficiency
♦ Conspicuous bronze color of the skin, especially in the creases of the hands and over the metacarpophalangeal joints (hand and finger), elbows, and knees caused by elevated levels of corticotropin and melanocyte-stimulating hormone
♦ Darkening of scars, areas of vitiligo (absence of pigmentation), and increased pigmentation of the mucous membranes, especially the buccal mucosa, due to decreased secretion of cortisol, causing simultaneous, excessive secretion of corticotropin and melanocyte-stimulating hormone by the pituitary gland
♦ Associated cardiovascular abnormalities, including orthostatic hypotension, decreased cardiac size and output, and weak, irregular pulse caused by mineralocorticoid deficiency
♦ Decreased tolerance for even minor stress because of glucocorticoid deficiency
♦ Fasting hypoglycemia due to decreased gluconeogenesis
♦ Craving for salty food due to decreased mineralocorticoid secretion (which normally causes salt retention)
Secondary hypofunction
♦ Similar to primary hypofunction, but without hyperpigmentation due to low corticotropin and melanocyte-stimulating hormone levels
♦ Possibly no hypotension and electrolyte abnormalities due to fairly normal aldosterone secretion
♦ Usually normal androgen secretion
Addisonian crisis
Signs and symptoms are related to the severe deficiencies of adrenal hormones and may include:
♦ profound weakness and fatigue
♦ nausea, vomiting, and dehydration
♦ hypotension
♦ high fever followed by hypothermia (occasionally).
Complications
♦ Hyperpyrexia
♦ Psychotic reactions
♦ Deficient or excessive steroid treatment
♦ Shock
♦ Profound hypoglycemia
♦ Ultimate vascular collapse, renal shutdown, coma, and death (if untreated)
Diagnosis
♦ Plasma cortisol level confirming adrenal insufficiency (corticotropin stimulation test to differentiate between primary and secondary adrenal hypofunction)
♦ Metyrapone test for suspicion of secondary adrenal hypofunction (oral or I.V. metyrapone blocks cortisol production and should stimulate the release of corticotropin from the hypothalamicpituitary system; in Addison’s disease, the hypothalamic-pituitary system responds normally and the plasma corticotropin level is high, but because the adrenal glands are destroyed, the plasma level of the cortisol precursor 11-deoxycortisol increases, as does the urinary 17-hydroxycorticosteroid level)
♦ Corticotropin stimulation test by I.V. administration of corticotropin, after baseline sampling for plasma cortisol and 24-hour urine cortisol levels (in adrenal hypofunction, plasma and urine cortisol levels fail to rise normally, in response to corticotropin; in secondary hypofunction, repeated doses of corticotropin over successive days produce a gradual increase in cortisol levels until normal values are reached).
In a patient with typical addisonian symptoms, the following laboratory findings strongly suggest acute adrenal insufficiency:
♦ a decreased plasma cortisol level (less than 10 mcg/dl in the morning; less in the evening)
♦ decreased serum sodium and fasting blood glucose levels
♦ increased serum potassium, calcium, and blood urea nitrogen levels
♦ elevated hematocrit; increased lymphocyte and eosinophil counts
♦ X-rays showing adrenal calcification if the cause is infectious.
Treatment
♦ Lifelong corticosteroid replacement, usually with cortisone or hydrocortisone, both of which have a mineralocorticoid effect (primary or secondary adrenal hypofunction)
♦ Oral fludrocortisone (Florinef), a synthetic mineralocorticoid, to prevent dangerous dehydration, hypotension, hyponatremia, and hyperkalemia (Addison’s disease)
♦ In adrenal crisis, I.V. bolus of hydrocortisone, then given I.M. or I.V. until the patient’s condition stabilizes.
With proper treatment, adrenal crisis usually subsides quickly; blood pressure stabilizes, and water and sodium levels return to normal. After the crisis, maintenance doses of hydrocortisone preserve physiologic stability.
Special considerations
 In adrenal crisis, monitor the vital signs carefully, watching especially for hypotension, volume depletion, and other signs of shock (decreased level of consciousness and urine output). Also watch for hyperkalemia before treatment and hypokalemia after treatment (from excessive mineralocorticoid effect) and for cardiac arrhythmias (may be caused by a serum potassium disturbance).
In adrenal crisis, monitor the vital signs carefully, watching especially for hypotension, volume depletion, and other signs of shock (decreased level of consciousness and urine output). Also watch for hyperkalemia before treatment and hypokalemia after treatment (from excessive mineralocorticoid effect) and for cardiac arrhythmias (may be caused by a serum potassium disturbance).♦ If the patient also has diabetes, check his blood glucose level periodically because steroid replacement may require adjustment of insulin dosage.
♦ Record weight and intake and output carefully because the patient may have volume depletion. Until onset of mineralocorticoid effect, force fluids to replace excessive fluid loss.
To manage the patient receiving maintenance steroid therapy:
♦ Arrange for a diet that maintains sodium and potassium balance.
♦ If the patient is anorexic, suggest six small meals per day to increase caloric intake. Ask the dietitian to provide a diet high in protein and carbohydrates. Keep a late-morning snack available in case the patient becomes hypoglycemic.
♦ Observe the patient receiving a steroid for cushingoid signs, such as fluid retention around the eyes and face. Watch for fluid and electrolyte imbalance, especially if the patient is receiving a mineralocorticoid. Monitor weight and check blood pressure to assess body fluid status. Remember, steroids administered in the late afternoon or evening may stimulate of the central nervous system and cause insomnia in some patients. Check for petechiae because these patients bruise easily.
♦ If the patient receives only a glucocorticoid, observe for orthostatic hypotension or abnormal electrolyte levels, which may indicate a need for mineralocorticoid therapy.
♦ Explain that lifelong steroid therapy is necessary.
♦ Teach the patient the signs and symptoms of steroid overdose (swelling, weight gain) and steroid underdose (lethargy, weakness).
♦ Tell the patient that his dose may need to be increased during times of stress (when he has a cold, for example).
♦ Warn that infection, injury, or profuse sweating in hot weather may precipitate adrenal crisis.
♦ Instruct the patient to always carry a medical identification card stating that he takes a steroid and giving the name of the drug and the dosage.
♦ Teach the patient and family how to give a hydrocortisone injection.
♦ Tell the patient to keep an emergency kit available containing hydrocortisone in a prepared syringe for use in times of stress.
♦ Warn the patient that stress may necessitate additional cortisone to prevent adrenal crisis.
CONGENITAL ADRENAL HYPERPLASIA
Congenital adrenal hyperplasia (CAH) encompasses a group of genetic disorders resulting in the deficiency or absence of one of five enzymes needed for the biosynthesis of glucocorticoids and mineralocorticoids. Manifestations are usually present at birth or during early childhood, but symptoms may appear later in life in nonclassic CAH. CAH is uncommon and typically has an autosomal recessive mode of inheritance. When successfully treated, sexual functioning and fertility aren’t affected.
The most common adrenal disorder in infants and children is CAH. There are two common forms: simple virilizing CAH and salt-losing CAH. Acquired adrenal virilism, usually the result of an adrenal tumor, is rare and affects twice as many females as males. With successful treatment, a normal quality of life and life span are expected. In older patients, androgen excess may be part of the syndrome of polycystic ovaries or may be secondary to an adrenal carcinoma or ovarian teratoma.
 Salt-losing CAH may cause fatal adrenal crisis in neonates.
Salt-losing CAH may cause fatal adrenal crisis in neonates.Causes
The cause of CAH is genetic, as an autosomal recessive trait.
Pathophysiology
The cortisol level is regulated by a negativefeedback mechanism. Corticotropin in the blood stimulates the release of cortisol precursors and, consequently, of cortisol, aldosterone, and androgens. In turn, cortisol suppresses corticotropin secretion. With a deficiency of the enzyme 21-hydroxylase, cortical secretion of cortisol is impaired and pituitary secretion of corticotropin is increased. Corticotropin stimulates the adrenal cortex, which in turn stimulates aldosterone and androgen biosynthesis and release.
In salt-losing CAH, 21-hydroxylase is almost absent. Corticotropin secretion increases, causing excessive production of cortisol precursors, including salt-wasting compounds. However, cortisol and aldosterone levels—both dependent on 21-hydroxylase—fall precipitously and, combined with the excessive production of saltwasting compounds, cause acute renal crisis. Corticotropin hypersecretion may stimulate adrenal androgens even more than in simple virilizing CAH, and produces masculinization.
Signs and symptoms
♦ Ambiguous genitalia (enlarged clitoris with urethral opening at the base and a combination of the labia and scrotum) caused by virilization from increased androgens, otherwise normal genital tract and gonads (female neonates)
♦ Pubic and axillary hair at an earlier age, a deep voice, acne, and facial hair, but no menarche (female approaching puberty), also resulting from an increased androgen level
♦ No apparent manifestations (male neonates)
♦ Accentuated masculine characteristics, including a deepened voice, acne, enlarged phallus with small testes, and frequent erections (male approaching puberty) resulting from increased androgen biosynthesis and release
♦ A high androgen level causing rapid bone and muscle growth (children)
♦ Short stature due to premature epiphyseal closure and a high androgen level (adults)
♦ More severe changes, including development of a penis in females (salt-losing CAH)
Because males have no external abnormalities, diagnosis is more difficult and commonly delayed until other signs occur. In the second week of life, signs of a salt-wasting crisis include apathy, failure to eat, diarrhea, and adrenal crisis (vomiting, dehydration from hyponatremia, and hyperkalemia). If adrenal crisis isn’t treated promptly, dehydration and electrolyte imbalance cause cardiovascular collapse and cardiac arrest.
Complications
♦ Death (salt-wasting crisis) due to dehydration and hyperkalemia
♦ Precocious puberty
♦ Menstrual irregularities
♦ Sexual dysfunction, infertility, and altered external genitalia
♦ Adrenal crisis
♦ Altered growth
Diagnosis
♦ An elevated urine 17-ketosteroid level (can be suppressed by dexamethasone [Decadron])
♦ An elevated serum 17-hydroxyprogesterone level after I.V. bolus of corticotropin
♦ Serum hyperkalemia, hyponatremia, and hypochloremia (present but not diagnostic)
♦ An elevated 24-hour urine pregnanetriol level
♦ Normal or decreased 24-hour urine 17-hydroxycorticosteroid levels
Treatment
♦ Daily cortisone (Cortone) or hydrocortisone (Cortef) to stop the excessive output of corticotropin and subsequent excessive androgen production (initial and subsequent doses guided by urinary 17-ketosteroid levels) given I.M. until the infant is old enough to tolerate pills (usually about 18 months)
♦ I.V. sodium chloride and glucose to reestablish and maintain fluid and electrolyte balance, with desoxycorticosterone I.M. and hydrocortisone I.V. as needed (adrenal crisis); glucocorticoid (cortisone or hydrocortisone) and perhaps mineralocorticoids (desoxycorticosterone, fludrocortisone, or both after stabilization)
♦ Sex chromatin and karyotype studies to determine genetic sex (with ambiguous external genitalia); possible reconstructive surgery for females between ages 1 and 3
Special considerations
Suspect CAH in infants hospitalized for failure to thrive, dehydration, or diarrhea as well as in tall, sturdy-looking children with a record of numerous episodic illnesses.
♦ When caring for an infant with adrenal crisis, keep the I.V. line patent, infuse fluids, and give a steroid, as ordered. Monitor body weight, blood pressure, urine output, and serum electrolyte levels carefully, especially sodium and potassium levels.
♦ Watch for signs of shock (cyanosis, hypotension, tachycardia, and tachypnea).
♦ If the child is receiving maintenance therapy with steroid injections, rotate I.M. injection sites to prevent atrophy; tell parents to do the same. Teach them the possible adverse effects (cushingoid symptoms) of long-term therapy. Explain that lifelong maintenance therapy with hydrocortisone, cortisone, or the mineralocorticoid fludrocortisone is essential for survival. Warn parents not to stop therapy with these drugs suddenly because potentially fatal adrenal hypofunction will result. Instruct parents to report stress and infection, which require increased steroid doses.
♦ Monitor the patient receiving desoxycorticosterone or fludrocortisone for edema, weakness, and hypertension. Be alert for significant weight gain and rapid changes in height because normal growth is an important indicator of adequate therapy.
♦ Instruct the patient to wear a medical identification bracelet indicating that he’s on prolonged steroid therapy and providing information about dosage.
♦ Help the parents of a female infant with male genitalia to understand that she’s physiologically a female and that this abnormality can be surgically corrected. Arrange for counseling if necessary.
CUSHING’S SYNDROME
Cushing’s syndrome is a cluster of clinical abnormalities caused by prolonged exposure to elevated levels of endogenous or exogenous glucocorticoids. Most cases in the United States result from exogenous glucocorticoids. Of the 13 cases per million that occur annually, 10% result from a pituitary adrenocorticotropic hormone (ACTH)-producing tumor.
Rare adrenal carcinomas are associated with a 5-year survival rate of less than 30%. Cushing’s syndrome that results from an adrenal or a pituitary tumor affects women five times more frequently than men, with a peak incidence between ages 25 and 40.
Causes
♦ ACTH-producing pituitary adenoma
♦ Adrenal adenoma, carcinoma, or hyperplasia
♦ Autonomous, ectopic corticotropin secretion by a tumor outside the pituitary (usually malignant, commonly oat cell carcinoma of the lung)
♦ Excessive glucocorticoid administration, including prolonged use
Pathophysiology
Cushing’s syndrome is caused by prolonged exposure to excess glucocorticoids. Cushing’s syndrome can be exogenous, resulting from chronic glucocorticoid or corticotropin administration, or endogenous, resulting from increased cortisol or corticotropin secretion. Cortisol excess results in anti-inflammatory effects and excessive catabolism of protein and peripheral fat to support hepatic glucose production. The mechanism may be corticotropin dependent (an elevated plasma corticotropin level stimulates the adrenal cortex to produce excess cortisol) or corticotropin independent (excess cortisol is produced by the adrenal cortex or exogenously administered). Excess cortisol suppresses the hypothalamicpituitary-adrenal axis, also present in ectopic corticotropin-secreting tumors.
Signs and symptoms
Like other endocrine disorders, Cushing’s syndrome induces changes in many body systems. Signs and symptoms depend on the degree and duration of hypercortisolism, the presence or absence of androgen excess, and additional tumor-related effects (adrenal carcinoma or ectopic corticotropin syndrome). Specific clinical effects vary with the system affected and include:
♦ diabetes mellitus, with decreased glucose tolerance, fasting hyperglycemia, and glycosuria due to cortisol-induced insulin resistance and increased gluconeogenesis in the liver (endocrine and metabolic systems)
♦ muscle weakness due to hypokalemia or loss of muscle mass due to increased catabolism, pathologic fractures due to decreased bone mineral ionization, osteopenia, osteoporosis,
and skeletal growth retardation in children (musculoskeletal system)
and skeletal growth retardation in children (musculoskeletal system)
♦ purple striae; facial plethora (edema and blood vessel distention); acne; fat pads above the clavicles, over the upper back (buffalo hump), on the face (moon facies), and around the trunk (truncal obesity) with slender arms and legs; little or no scar formation; poor wound healing due to decreased collagen and weakened tissues; spontaneous ecchymosis; hyperpigmentation; fungal skin infections (integumentary system)
♦ peptic ulcer due to increased gastric secretions and pepsin production and decreased gastric mucus, abdominal pain, increased appetite, weight gain (GI system)
♦ irritability and emotional lability, ranging from euphoric behavior to depression or psychosis; insomnia due to the cortisol’s role in neurotransmission; headache (central nervous system [CNS])
♦ hypertension due to sodium and secondary fluid retention; heart failure; left ventricular hypertrophy; capillary weakness from protein loss, which leads to bleeding and ecchymosis; dyslipidemia; ankle edema (cardiovascular system)
♦ increased susceptibility to infection due to decreased lymphocyte production and suppressed antibody formation; decreased resistance to stress; suppressed inflammatory response masking even severe infection (immunologic system)
♦ fluid retention, increased potassium excretion, ureteral calculi from increased bone demineralization with hypercalciuria (renal and urologic systems)
♦ increased androgen production with clitoral hypertrophy, mild virilism, hirsutism, and amenorrhea or oligomenorrhea in women; sexual dysfunction; decreased libido; impotence (reproductive system).
Complications
♦ Osteoporosis
♦ Increased susceptibility to infections
♦ Hirsutism
♦ Ureteral calculi
♦ Metastasis of malignant tumors
Diagnosis
♦ Hyperglycemia, hypernatremia, glycosuria, hypokalemia, and metabolic alkalosis
♦ Urinary free-cortisol levels more than 150 mcg/24 hours
♦ Dexamethasone suppression test to confirm the diagnosis and determine the cause, possibly an adrenal tumor or a nonendocrine, corticotropinsecreting tumor
♦ Elevated serum cortisol levels when sample taken at 11 p.m. (early finding)
♦ Salivary cortisol levels greater than 1.3 ng/ml (radioimmunoassay) or greater than 1.5 ng/ml (competitive-protein binding assay)
♦ Blood levels of corticotropin-releasing hormone, corticotropin, and different glucocorticoids to diagnose and localize cause to pituitary or adrenal gland
♦ Radiologic evaluation using ultrasonography, computed tomography scan, or magnetic resonance imaging enhanced with gadolinium to locate a causative tumor in the pituitary or adrenal glands
♦ Petrosal sinus sampling to determine whether Cushing’s syndrome is due to a pituitary tumor or some other cause
Treatment
Differentiation among pituitary, adrenal, and ectopic causes of hypercortisolism is essential for effective treatment, which is specific for the cause of cortisol excess and includes medication, radiation, and surgery. Possible treatments include:
♦ surgery for tumors of the adrenal and pituitary glands or other tissue (such as the lung)
♦ radiation therapy (tumor)
♦ drug therapy, which may include ketoconazole, metyrapone, and aminoglutethimide to inhibit cortisol synthesis; mitotane to destroy the adrenocortical cells that secret cortisol; and bromocriptine and cyproheptadine to inhibit corticotropin secretion.
Special considerations
Patients with Cushing’s syndrome require painstaking assessment and vigorous supportive care:
♦ Frequently monitor vital signs, especially blood pressure. Carefully observe the hypertensive patient who also has cardiac disease.
♦ Check laboratory reports for hypernatremia, hypokalemia, hyperglycemia, and glycosuria.
♦ Because the cushingoid patient is likely to retain sodium and water, check for edema and monitor daily weight and intake and output carefully. To minimize weight gain, edema, and hypertension, ask the dietary department to provide a diet that’s high in protein and potassium but low in calories, carbohydrates, and sodium.
♦ Watch for infection, which is a particular problem in Cushing’s syndrome.
♦ If the patient has osteoporosis and is bedridden, perform passive range-of-motion exercises carefully because of the severe risk of pathologic fractures.
♦ Remember, Cushing’s syndrome produces emotional lability. Record incidents that upset the patient, and try to prevent such situations from occurring, if possible. Help him get the physical and mental rest he needs—using sedation, if necessary. Offer support to the emotionally labile patient throughout the difficult testing period.
After bilateral adrenalectomy and pituitary surgery:
♦ Be sure to report wound drainage or temperature elevation to the patient’s physician immediately. Use strict sterile technique in changing the patient’s dressings.
♦ Administer an analgesic and a replacement steroid as ordered.
♦ Monitor urine output and check vital signs carefully, watching for signs of shock (decreased blood pressure, increased pulse rate, pallor, and cold, clammy skin). To counteract shock, give a vasopressor and increase the rate of I.V. fluids, as ordered. Because mitotane, aminoglutethimide, and metyrapone decrease mental alertness and produce physical weakness, assess neurologic and behavioral status, and warn the patient of adverse CNS effects. Also, watch for severe nausea, vomiting, and diarrhea.
♦ Check laboratory reports for hypoglycemia due to removal of the source of cortisol, a hormone that maintains the blood glucose level.
♦ Check for abdominal distention and return of bowel sounds after adrenalectomy.
♦ Check regularly for signs and symptoms of adrenal hypofunction (orthostatic hypotension, apathy, weakness, fatigue), which indicate that steroid replacement is inadequate.
♦ In the patient undergoing pituitary surgery, check for and immediately report signs and symptoms of increased intracranial pressure (confusion, agitation, changes in level of consciousness, nausea, and vomiting). Watch for hypopituitarism.
Provide comprehensive teaching to help the patient cope with lifelong treatment:
♦ Advise the patient to take the replacement steroid with an antacid or a meal, to minimize gastric irritation. (Usually, it’s helpful to take two-thirds of the dose in the morning and the remaining third in the early afternoon to mimic diurnal adrenal secretion.)
♦ Tell the patient to carry a medical identification card and to immediately report physiologically stressful situations, such as infections, which necessitate an increased dose.
♦ Instruct the patient to watch closely for signs and symptoms of inadequate steroid dosing (fatigue, weakness, dizziness) and of overdosage (severe edema, weight gain). Emphatically warn against abrupt discontinuation of steroid therapy because this may produce a fatal adrenal crisis.
DIABETES INSIPIDUS
A disorder of water metabolism, diabetes insipidus results from a deficiency of circulating vasopressin (also called antidiuretic hormone, or ADH) or from renal resistance to this hormone. Pituitary diabetes insipidus is caused by a deficiency of vasopressin, and nephrogenic diabetes insipidus is caused by the resistance of renal tubules to vasopressin. Diabetes insipidus is characterized by excessive fluid intake and hypotonic polyuria. A decrease in the ADH level leads to altered intracellular and extracellular fluid control, causing renal excretion of a large amount of urine.
The disorder may start at any age and is slightly more common in men than in women. The incidence is slightly greater today than in the past.
In uncomplicated diabetes insipidus, the prognosis is good with adequate water replacement, and patients usually lead normal lives.
Causes
♦ Acquired, familial, idiopathic, neurogenic, or nephrogenic
♦ Associated with stroke, hypothalamic or pituitary tumors, and cranial trauma or surgery (neurogenic diabetes insipidus)
♦ Certain drugs, such as lithium (Duralith), phenytoin (Dilantin), or alcohol (transient diabetes insipidus)
♦ X-linked recessive trait or end-stage renal failure (nephrogenic diabetes insipidus, less common)
Pathophysiology
Diabetes insipidus is related to an insufficiency of ADH, leading to polyuria and polydipsia. The three forms of diabetes insipidus are neurogenic, nephrogenic, and psychogenic.
Neurogenic, or central, diabetes insipidus is an inadequate response of ADH to plasma osmolarity, which occurs when an organic lesion of the hypothalamus, infundibular stem, or posterior pituitary partially or completely blocks ADH synthesis, transport, or release. Many organic lesions can cause diabetes insipidus, including brain tumors, hypophysectomy, aneurysms, thrombosis, skull fractures, infections, and immunologic disorders. Neurogenic diabetes insipidus has an acute onset. A three-phase syndrome can occur, which involves:
♦ progressive loss of nerve tissue and increased diuresis
♦ normal diuresis
♦ polyuria and polydipsia, the manifestation of permanent loss of the ability to secrete adequate ADH.
Nephrogenic diabetes insipidus is caused by an inadequate renal response to ADH. The collecting duct permeability to water doesn’t increase in response to ADH. Nephrogenic diabetes insipidus is generally related to disorders and drugs that damage the renal tubules or inhibit the generation of cyclic adenosine monophosphate in the tubules, preventing activation of the second messenger. Causative disorders include pyelonephritis, amyloidosis, destructive uropathies, polycystic disease, and intrinsic renal disease. Drugs include lithium (Eskalith); general anesthetics, such as methoxyflurane; and demeclocycline (Declomycin). In addition, hypokalemia or hypercalcemia impairs the renal response to ADH. A rare genetic form of nephrogenic diabetes insipidus is an X-linked recessive trait.
Psychogenic diabetes insipidus is caused by excessive fluid intake, which may be idiopathic or related to psychosis or sarcoidosis. The polydipsia and resultant polyuria wash out ADH more quickly than it can be replaced. Chronic polyuria may overwhelm the renal medullary concentration gradient, rendering patients partially or totally unable to concentrate urine.
Regardless of the cause, insufficient ADH causes the immediate excretion of large volumes of dilute urine and consequent plasma hyperosmolality. In conscious individuals, the thirst mechanism is stimulated, usually for cold liquids. With severe ADH deficiency, urine output may be greater than 12 L/day, with a low specific gravity. Dehydration develops rapidly if fluids aren’t replaced.
Signs and symptoms
♦ Polydipsia (cardinal sign)—fluid intake caused by stimulation of the thirst mechanism
♦ Polyuria (cardinal sign)—urine output of 4 to 16 L/24-hour period of dilute urine caused by insufficient ADH
♦ Nocturia—caused by increased urine output leading to sleep disturbance and fatigue
♦ Low urine specific gravity—less than 1.006 caused by polyuria
♦ Changes in level of consciousness caused by central nervous system cellular dehydration
♦ Hypotension and tachycardia caused by a decrease in vascular volume related to fluid loss
♦ Headache and visual disturbance due to electrolyte disturbance and dehydration
♦ Abdominal fullness, anorexia, and weight loss due to almost continuous fluid consumption
Complications
♦ Dilation of the urinary tract
♦ Severe dehydration
♦ Shock and renal failure if dehydration is severe
Diagnosis
♦ Urinalysis showing almost colorless urine of low osmolality (50 to 200 mOsm/kg, less than that of plasma) and low specific gravity (less than 1.005)
♦ Water deprivation test to identify vasopressin deficiency, resulting in renal inability to concentrate urine
Treatment
Until the cause of diabetes insipidus can be identified and eliminated, the administration of vasopressin (Pitressin) can control fluid balance and prevent dehydration. Medications include:
♦ hydrochlorothiazide with potassium supplement for central and nephrogenic diabetes insipidus
♦ vasopressin aqueous preparation subcutaneously several times daily, effective for only 2 to 6 hours (used as a diagnostic agent and, rarely, in acute disease)
♦ desmopressin acetate (DDAVP) orally, by nasal spray absorbed through the mucous membranes, or by subcutaneous or I.V. injection, effective for 8 to 20 hours depending on the dose
♦ indomethacin and amiloride for nephrogenic diabetes insipidus.
Special considerations
♦ Patient care includes monitoring symptoms to ensure that fluid balance is restored and maintained.
♦ Record fluid intake and output carefully. Maintain adequate fluid intake to prevent severe dehydration. Watch for signs of hypovolemic shock, and monitor blood pressure and heart and respiratory rates regularly, especially during the water deprivation test. Check the patient’s weight daily.
♦ If the patient is dizzy or has muscle weakness, keep the side rails up and assist him with walking.
♦ Monitor urine specific gravity between doses. Watch for a decrease in specific gravity accompanied by increased urine output, indicating the recurrence of polyuria and necessitating administration of the next dose of medication or a dosage increase.
♦ Monitor serum electrolyte levels closely. Report abnormal values, and provide treatment as ordered.
♦ If constipation develops, add more high-fiber foods and fruit juices to the patient’s diet. If necessary, obtain an order for a mild laxative, such as milk of magnesia.
♦ Provide meticulous skin and mouth care; apply petroleum jelly as needed to cracked or sore lips.
♦ Before discharge, teach the patient how to monitor intake and output.
♦ Instruct the patient to administer desmopressin by nasal spray only after the onset of polyuria—not before—to prevent excess fluid retention and water intoxication.
♦ Tell the patient to report weight gain, which may indicate that his medication dose is too high. Recurrence of polyuria, as reflected on the intake and output sheet, indicates that the dose is too low.
♦ Teach the parents of a child with diabetes insipidus about normal growth and development. Discuss how their child may differ from others at his developmental stage.
♦ Advise the patient with diabetes insipidus to wear a medical identification bracelet and to carry his medication with him at all times.
DIABETES MELLITUS
Diabetes mellitus is a metabolic disorder characterized by hyperglycemia (an elevated blood glucose level) resulting from lack of insulin, lack of insulin effect, or both. Two general classifications are recognized:
♦ type 1, absolute insulin insufficiency
♦ type 2, insulin resistance with varying degrees of insulin secretory defects.
Several secondary forms also exist, caused by such conditions as pregnancy (gestational diabetes mellitus), pancreatic disease, hormonal or genetic problems, and certain drugs or chemicals.
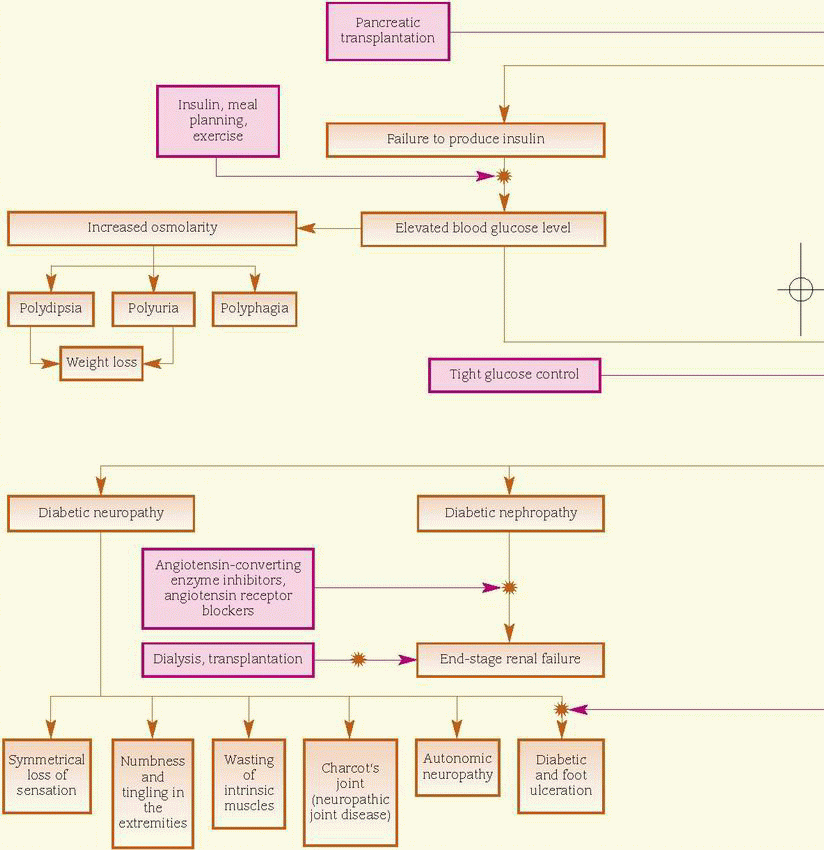
Onset of type 1 (insulin-dependent) usually occurs before age 30 (although it may occur at any age); the patient is usually thin and requires exogenous insulin and dietary management to achieve control. (See Treatment of type 1 diabetes mellitus, pages 476 and 477.) Conversely, type 2 (non-insulin-dependent) usually occurs in obese adults after age 40 and is treated with diet and exercise in combination with various oral antidiabetics, although treatment may include insulin therapy.
Medical advances permit increased longevity and improved quality of life if the patient carefully monitors the blood glucose level, uses the data to make pharmacologic and lifestyle changes, and uses new insulin delivery systems, such as subcutaneous insulin pumps. In addition, medications now available enhance the body’s own glucose metabolism and insulin sensitivity to optimize glycemic control and prevent progression to long-term complications.
Causes
♦ Conditions that antagonize the actions of insulin (Cushing’s syndrome, acromegaly, pheochromocytoma)
♦ Environment (infection, diet, toxins, stress, glucocorticoids)
♦ Heredity
♦ Lifestyle changes in genetically susceptible persons
♦ Pregnancy
Pathophysiology
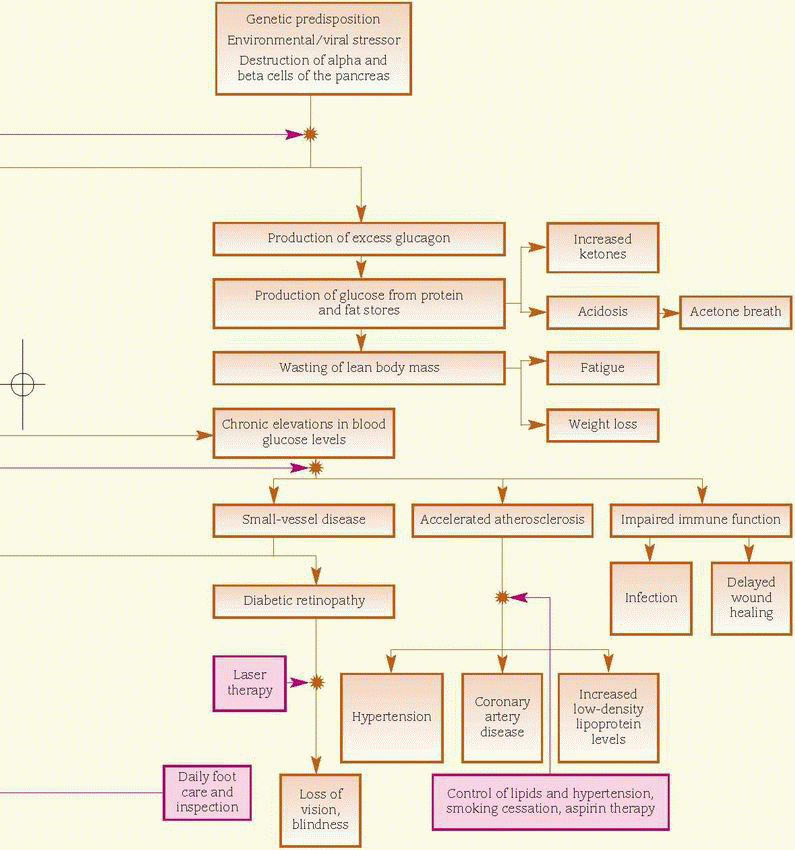
In persons genetically susceptible to type 1 diabetes, a triggering event, possibly a viral infection, causes production of autoantibodies against the beta cells of the pancreas. The resultant destruction of the beta cells leads to a decline in and ultimate lack of insulin secretion. Insulin deficiency leads to hyperglycemia, enhanced lipolysis (decomposition of fat), and protein catabolism. These characteristics occur when more than 90% of the beta cells have been destroyed.
Type 2 diabetes mellitus is a chronic disease caused by one or more of the following factors: impaired insulin secretion, inappropriate hepatic glucose production, or peripheral insulin receptor insensitivity. Genetic factors are significant for both pancreatic beta-cell failure and insulin resistance, and onset is accelerated by obesity and a sedentary lifestyle. Again, added stress can be a pivotal factor. (See Understanding type 2 diabetes, page 478, and How diabetes mellitus affects the body, page 479.)
Gestational diabetes mellitus occurs when a woman not previously diagnosed with diabetes shows glucose intolerance during pregnancy. This may occur if placental hormones counteract insulin, causing insulin resistance. Gestational diabetes mellitus is a significant risk factor for the future occurrence of type 2 diabetes mellitus.
Signs and symptoms
 Type 1 diabetes usually presents rapidly, typically with polydipsia, polyuria, polyphagia, weakness, weight loss, dry skin, and ketoacidosis. Type 2 diabetes is typically slow and insidious in onset and usually unaccompanied by symptoms.
Type 1 diabetes usually presents rapidly, typically with polydipsia, polyuria, polyphagia, weakness, weight loss, dry skin, and ketoacidosis. Type 2 diabetes is typically slow and insidious in onset and usually unaccompanied by symptoms.♦ Polyuria and polydipsia due to high serum osmolality caused by a high blood glucose level
♦ Anorexia (common) resulting from an elevated blood glucose level or polyphagia (occasional)
most likely caused by cellular starvation and cellular depletion of nutrient stores
most likely caused by cellular starvation and cellular depletion of nutrient stores
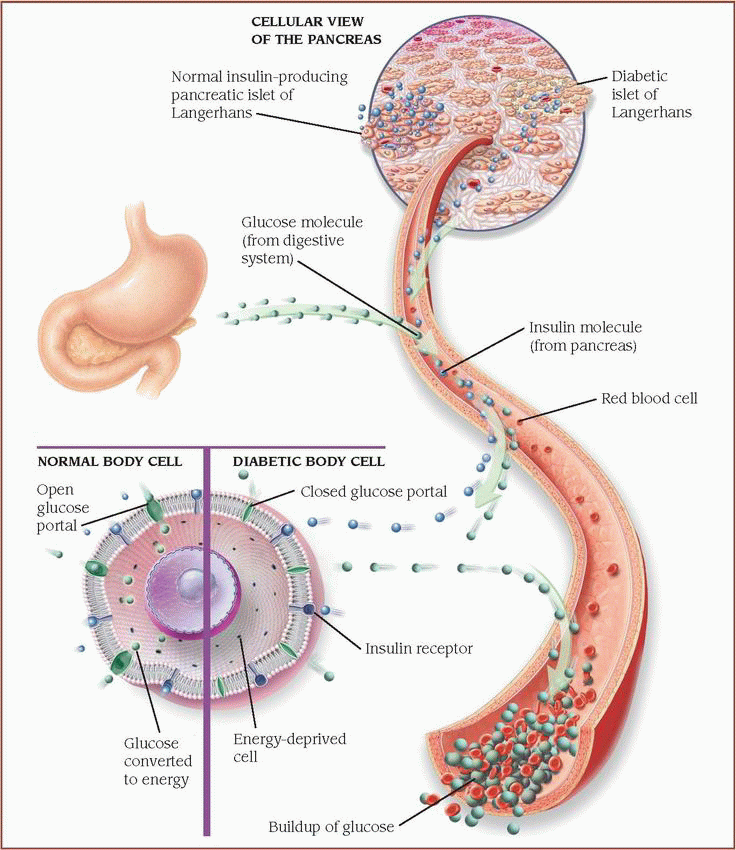
Normally, in response to blood glucose levels, the pancreatic islets of Langerhans release insulin. In type 2 diabetes, problems arise when insufficient insulin is produced or when the body’s cells resist insulin.
Normal body cell
Normally, insulin molecules bind to the preceptors on the body’s cells. When activated by insulin, portals open to allow glucose to enter the cell, where it’s converted to energy.
Diabetes mellitus can affect most major body systems. For this reason, a team approach to management and care is needed.
Cardiovascular system
♦ Arterial thrombosis may develop due to persistent activated thrombogenic pathways and impaired fibrinolysis.
GI system
♦ Autonomic neuropathy leads to abdominal discomfort and pain, causing gastroparesis and constipation.
♦ Nausea, diarrhea, or constipation may develop due to dehydration, electrolyte imbalances, or autonomic neuropathy.
Metabolic system
♦ Impaired or absent insulin function prevents normal metabolism of carbohydrates, fats, and proteins, leading to weight loss.
♦ Muscle cramps, irritability, and emotional lability result from electrolyte imbalances.
Neurologic system
♦ A low intracellular glucose level may result in headaches, fatigue, lethargy, reduced energy levels, and impaired school and work performance.
♦ Vision changes, such as blurring, occur due to glucose-induced swelling.
♦ Numbness and tingling occur due to neural tissue damage.
Renal system
♦ Polyuria and polydipsia occur because of high serum osmolality caused by a high serum glucose level.
Collaborative management
The patient may be managed by an endocrinologist to help control the blood glucose level, rehydrate him, and restore electrolyte and acid-base balance. Depending on the severity of the symptoms, the patient may require a pulmonologist to assist with ventilatory support. Nutritional therapy is indicated to assist with dietary needs. A diabetes educator can be valuable in helping the patient learn about his disease and how to manage it. Social services may assist with identifying financial and community resources and with arranging follow-up care.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


