Chapter 19 Endocrine disease
Introduction
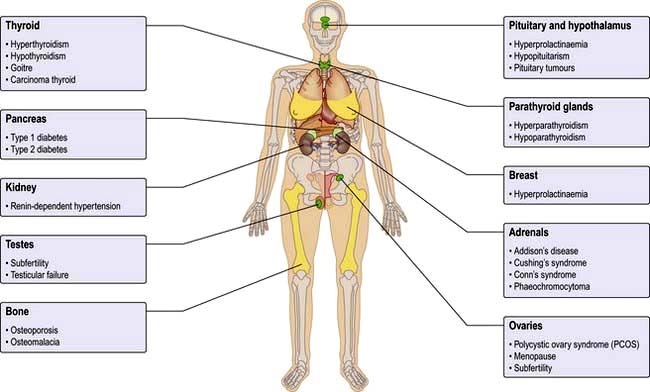
The endocrine system consists of glands that exert their actions at distant parts of the body via the production of biologically active hormones secreted into the bloodstream. Unlike the neurological system, which produces an immediate response, the endocrine system typically has a slower and longer lasting effect on the body. The main endocrine glands are the pituitary, thyroid, adrenals, gonads, parathyroids and pancreas and the common endocrine problems seen in clinical practice are shown in Figure 19.1. The pituitary gland, a pea-sized structure situated at the base of the brain, plays a key role in the control and feedback mechanisms of the endocrine system and has been termed the ‘conductor of the endocrine orchestra’.
Clinical presentation of endocrine disease
History
 The past history should include previous surgery or radiation involving endocrine glands, menstrual history, pregnancy and growth and development in childhood.
The past history should include previous surgery or radiation involving endocrine glands, menstrual history, pregnancy and growth and development in childhood.
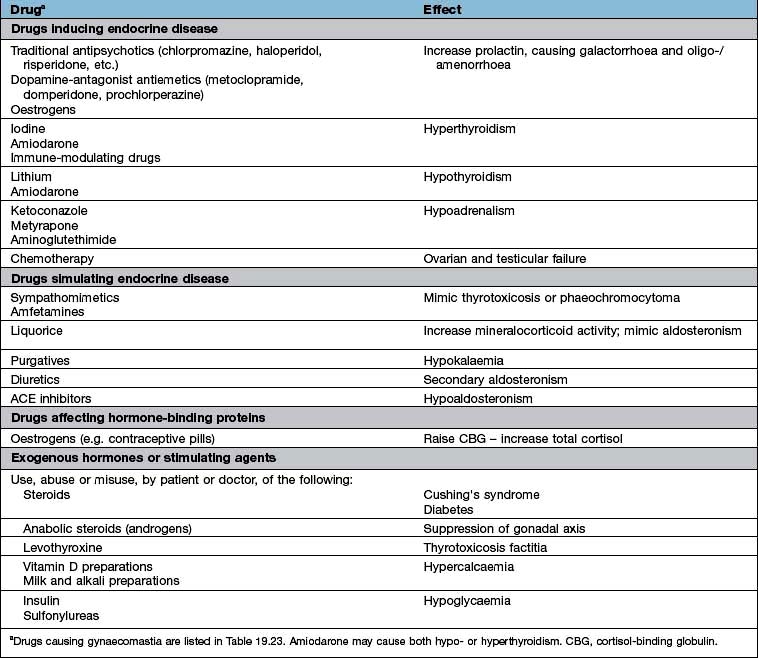
 A full drug history will exclude common iatrogenic endocrine problems (Table 19.1).
A full drug history will exclude common iatrogenic endocrine problems (Table 19.1).

Table 19.1 Drugs and endocrine disease
 |
Common endocrine conditions
The most common endocrine disorders, excluding obesity (Ch. 5) and diabetes mellitus (Ch. 20), are:
 Thyroid disorders: hypothyroidism, hyperthyroidism, goitre
Thyroid disorders: hypothyroidism, hyperthyroidism, goitre

 Hirsutism, usually due to polycystic ovary syndrome
Hirsutism, usually due to polycystic ovary syndrome

 Osteoporosis and metabolic bone disease
Osteoporosis and metabolic bone disease

Aetiology of endocrine disease
Aetiological mechanisms common to many endocrine disorders include:
Autoimmune disease
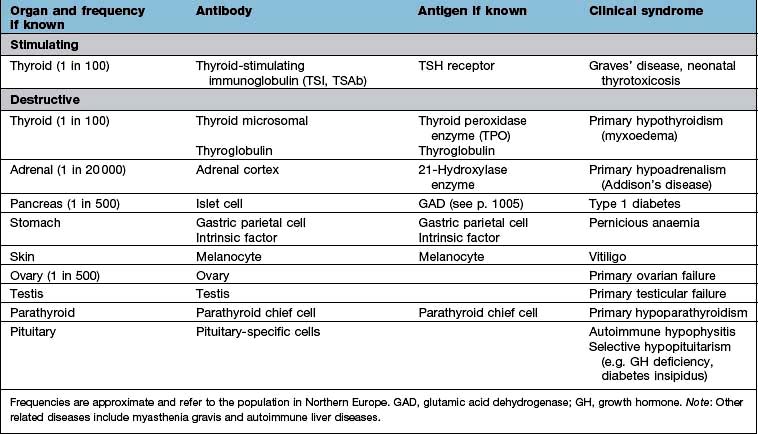
Organ-specific autoimmune diseases can affect every major endocrine organ (Table 19.2). They are characterized by the presence of specific antibodies in the serum, often present years before clinical symptoms are evident, are usually more common in women and have a strong genetic component, often with an identical-twin concordance rate of 50% and with HLA associations (see individual diseases). Several of the autoantigens have been identified.
Endocrine tumours
Most endocrine tumours are benign, although a cytological or histological diagnosis may be needed if there is clinical or radiological suspicion of malignancy. Clinical presentation depends on whether the tumour is functional or non-functional, the latter presenting only as a mass clinically or on imaging. Palpable thyroid nodules are common, and mass effects are a frequent presentation of pituitary adenomas, but the increased use of high-resolution ultrasound and detailed cross-sectional imaging has revealed a very high prevalence of asymptomatic, incidentally-discovered thyroid, adrenal and pituitary lesions, commonly termed ‘incidentalomas’ (see p. 989).
Endocrine adenomas typically present in a single gland, although rarer multiple endocrine neoplasia (MEN) syndromes exist due to very specific mutations of a single gene, such as the mutations of the RET proto-oncogene in MEN 2 or the MEN1 gene mutation in MEN 1 (see p. 997).
Enzyme defects
The biosynthesis of most hormones involves many stages. Deficient or abnormal enzymes can lead to absent or reduced production of the secreted hormone. In general, severe deficiencies present early in life with obvious signs; partial deficiencies usually present later with mild signs or are only evident under stress. An example of an enzyme deficiency is congenital adrenal hyperplasia (CAH), where the molecular basis has also been identified as mutations or deletions of the gene encoding the relevant enzymes (see p. 987).
Receptor abnormalities
There are rare conditions in which hormone secretion and control are normal but the receptors are defective; thus, if androgen receptors are defective, normal levels of androgen will not produce masculinization (e.g. testicular feminization). There are also a number of rare syndromes of diabetes and insulin resistance from receptor abnormalities (see p. 1006); other examples include nephrogenic diabetes insipidus, pseudohypoparathyroidism and thyroid hormone resistance which can cause an unusual pattern of thyroid blood results.
Hormonal activity
Hormone action and receptors
Hormone receptors are broadly divided into:
 Cell surface or membrane receptors: typically transmembrane receptors which contain hydrophobic sections spanning the lipid-rich plasma membrane and which trigger internal cellular messengers (see also p. 18)
Cell surface or membrane receptors: typically transmembrane receptors which contain hydrophobic sections spanning the lipid-rich plasma membrane and which trigger internal cellular messengers (see also p. 18)
 Nuclear receptors which typically bind hormones and translocate them to the nucleus where they bind hormone response elements of nuclear DNA via characteristic amino-acid sequences (e.g. so-called ‘zinc fingers’, see p. 27).
Nuclear receptors which typically bind hormones and translocate them to the nucleus where they bind hormone response elements of nuclear DNA via characteristic amino-acid sequences (e.g. so-called ‘zinc fingers’, see p. 27).
Abnormal receptors are an occasional cause of endocrine disease (see p. 43).
Mechanisms of hormone-receptor action
Common structural mechanisms of hormone-receptor action are illustrated in Figure 2.9 (p. 25) and include:
 stimulate cyclic AMP (cAMP) generation by adenylate cyclase – activating further intracellular kinases and leading to phosphorylation
stimulate cyclic AMP (cAMP) generation by adenylate cyclase – activating further intracellular kinases and leading to phosphorylation

 lead to diacylglycerol (DAG) activation of C-kinase and subsequent protein phosphorylation.
lead to diacylglycerol (DAG) activation of C-kinase and subsequent protein phosphorylation.
Most peptide hormones act via G-protein coupled receptors.
 Stimulation or release of pre-formed hormone from storage granules
Stimulation or release of pre-formed hormone from storage granules
 Stimulation or synthesis of hormone and other cellular components
Stimulation or synthesis of hormone and other cellular components


Control and feedback
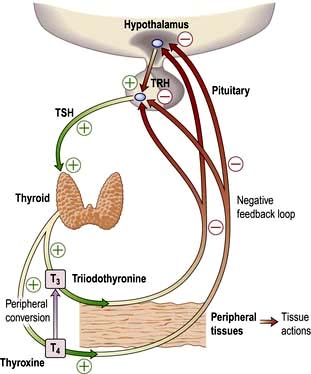
Most hormone systems are under tight regulatory control (typically by the hypothalamo-pituitary (HP) axis) by a system known as negative feedback. An example of the negative feedback system in the hypothalamo-pituitary-thyroid axis is demonstrated in Figure 19.2 and described here:
 TRH (thyrotrophin-releasing hormone) is secreted in the hypothalamus and travels via the portal system to the pituitary where it stimulates the thyrotrophs to produce thyroid-stimulating hormone (TSH).
TRH (thyrotrophin-releasing hormone) is secreted in the hypothalamus and travels via the portal system to the pituitary where it stimulates the thyrotrophs to produce thyroid-stimulating hormone (TSH).




 Peripheral T3 and T4 levels fall to normal.
Peripheral T3 and T4 levels fall to normal.


Primary and secondary gland failure
 ‘Primary’ hormone deficiency due to a disease process in the endocrine end-organ (thyroid, adrenal or gonad) will lead to a loss of negative feedback and subsequent elevation in the corresponding anterior pituitary hormone. Conversely, an abnormal hormone excess due to a disease process in the primary endocrine gland, or excess amount of exogenous hormone, will lead to increased negative feedback and suppression of the corresponding pituitary hormones.
‘Primary’ hormone deficiency due to a disease process in the endocrine end-organ (thyroid, adrenal or gonad) will lead to a loss of negative feedback and subsequent elevation in the corresponding anterior pituitary hormone. Conversely, an abnormal hormone excess due to a disease process in the primary endocrine gland, or excess amount of exogenous hormone, will lead to increased negative feedback and suppression of the corresponding pituitary hormones.

Measurement of hormones
Hormones are measured in routine clinical practice by biochemical assays in the laboratory. It is possible to measure pituitary trophic hormones and the hormones produced by the end-organ glands, but hypothalamic hormones are not routinely measured in practice because of their low concentration and local action within the hypothalamo-pituitary axis. Circulating levels of most hormones are very low (10−9– 10−12 mol/L) and cannot be measured by simple chemical techniques. Hormones are therefore usually measured by immunoassays, which rely on highly specific polyclonal or monoclonal antibodies, which bind to the hormone being measured during the assay incubation. This hormone-antibody interaction is measured by use of labelled hormone after separation of bound and free fractions (Fig. 19.3).
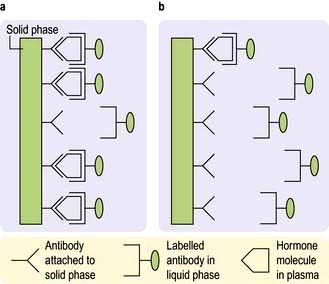
Hormone binding proteins
Many hormones are transported in the bloodstream from the primary gland to their distant target organ attached to a specific binding protein (p. 940). It is more helpful to measure the free hormone rather than total bound hormone level, as this is the part that is biologically active. Some modern assays attempt to measure the free hormone level directly (e.g. free T4) and are therefore a more accurate reflection of biological activity, although there are often technical problems with this approach and many assays still measure total hormone level.
Patterns of hormonal secretion
Hormone secretion can be continuous or intermittent, for example:
 Continuous secretion is shown by the thyroid hormones, with a half-life of 7–10 days for T4 and 6–10 hours for T3, and with little variation in levels over the day, month and year
Continuous secretion is shown by the thyroid hormones, with a half-life of 7–10 days for T4 and 6–10 hours for T3, and with little variation in levels over the day, month and year

Biological rhythms
Circadian means changes over the 24 hours of the day–night cycle and is best shown for the pituitary–adrenal axis. Figure 19.4 shows plasma cortisol levels measured over 24 hours – levels are highest in the early morning and lowest overnight. Additionally, cortisol release is pulsatile, following the pulsatility of pituitary ACTH. Thus ‘normal’ cortisol levels vary during the day and great variations can be seen in samples taken only 30 minutes apart.
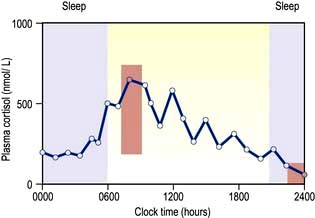
The menstrual cycle is an example of a longer and more complex (28-day) biological rhythm (see p. 972).



Testing endocrine function
Basal blood levels
Assays for all clinically relevant pituitary and end-organ hormones are available.
Stimulation and suppression tests
For example, where the secretory capacity of a gland is damaged, maximal stimulation by the trophic hormone will give a diminished output. Thus, in the short ACTH stimulation test for adrenal reserve (Box 19.1, Fig. 19.5a), the healthy subject shows a normal response while the subject with primary hypoadrenalism (Addison’s disease) demonstrates an impaired cortisol response to tetracosactide (an ACTH analogue).
 Box 19.1
Box 19.1






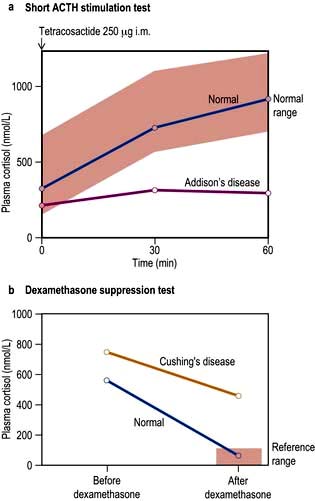
A patient with a hormone-producing tumour usually fails to show normal negative feedback. A patient with Cushing’s disease (excess pituitary ACTH) will thus fail to suppress ACTH and cortisol production when given a dose of synthetic steroid, in contrast to normal subjects. Figure 19.5b shows the response of a normal subject given dexamethasone 1 mg at midnight; cortisol is suppressed the following morning. The subject with Cushing’s disease shows inadequate suppression.
The pituitary gland and hypothalamus
Anatomy
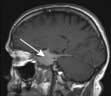
The anatomical relations of the hypothalamus and pituitary (Fig. 19.6) include the optic chiasm just above the pituitary fossa; any expanding lesion from the pituitary or hypothalamus can thus produce visual field defects by pressure on the chiasm. Such upward expansion of the gland through the diaphragma sellae is termed ‘suprasellar extension’. Lateral extension of pituitary lesions may involve the vascular and nervous structures in the cavernous sinus and may rarely reach the temporal lobe of the brain. The pituitary is itself encased in a bony box, therefore any lateral, anterior or posterior expansion must cause bony erosion.
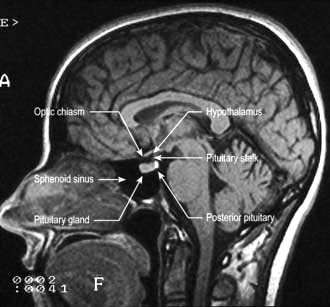
Figure 19.6 MR image of a sagittal section of the brain, showing the pituitary fossa and adjacent structures.
(By kind permission of Dr Martin Jeffree.)
Physiology
Hypothalamus
Hypothalamic neurones secrete pituitary hormone-releasing and -inhibiting factors and hormones (Table 19.3) into the portal system which run down the stalk to the pituitary. As well as the classical hormones illustrated in Figure 19.7, the hypothalamus also contains large amounts of other neuropeptides and neurotransmitters such as neuropeptide Y, vasoactive intestinal peptide (VIP) and nitric oxide that can also alter pituitary hormone secretion.
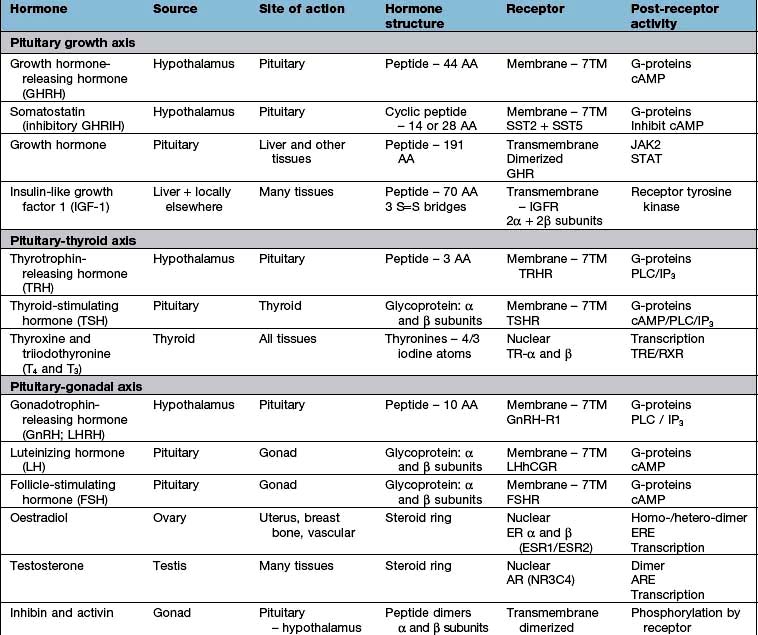
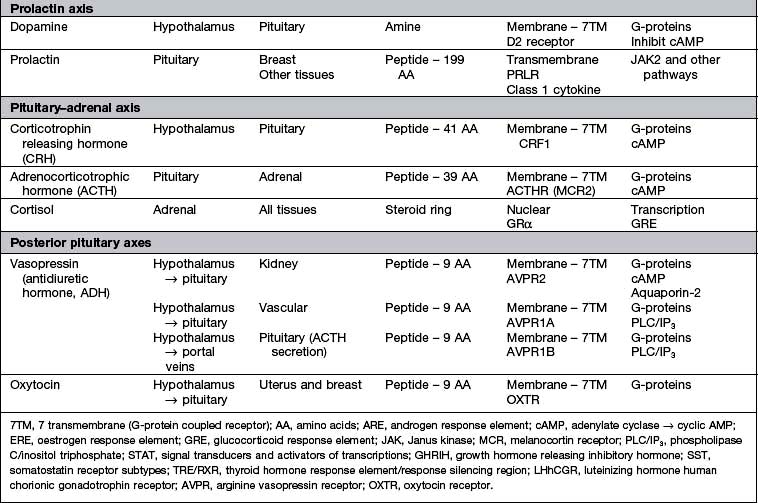
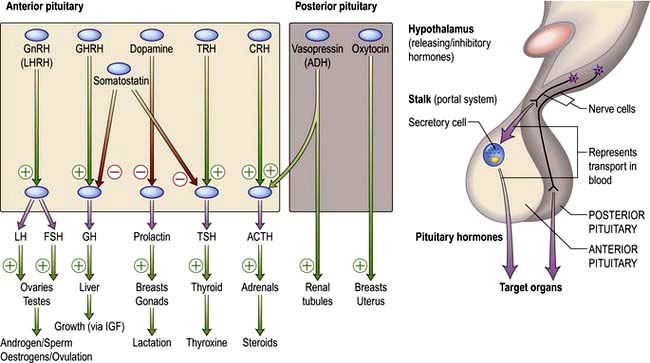
Figure 19.7 Hypothalamic releasing hormones and the pituitary trophic hormones. See the text for an explanation.
Posterior pituitary
The posterior pituitary is neuro-anatomically connected to specific hypothalamic nuclei, and acts merely as a storage organ. Antidiuretic hormone (ADH, also called vasopressin) and oxytocin, both nonapeptides, are synthesized in the supraoptic and paraventricular nuclei in the anterior hypothalamus. They are then transported along the axon and stored in the posterior pituitary (Fig. 19.7). This means that damage to the stalk or pituitary alone does not prevent synthesis and release of ADH and oxytocin. ADH is discussed on page 991; oxytocin produces milk ejection and uterine myometrial contraction.
Presentations of pituitary and hypothalamic disease
Pituitary space-occupying lesions and tumours
Pituitary tumours (Table 19.4) are the most common cause of pituitary disease, and the great majority of these are benign pituitary adenomas, usually monoclonal in origin. Problems are caused by:



Table 19.4 Characteristics of common pituitary and related tumours
| Tumour or condition | Usual size | Most common clinical presentation |
|---|---|---|
Prolactinoma | Most <10 mm (microprolactinoma) | Galactorrhoea, amenorrhoea, hypogonadism, erectile dysfunction |
| Some >10 mm (macroprolactinoma) | As above plus headaches, visual field defects and hypopituitarism |
Acromegaly | Few mm to several cm | Change in appearance, visual field defects and hypopituitarism |
Cushing’s disease | Most small: few mm (some cases are hyperplasia) | Central obesity, cushingoid appearance (local symptoms rare) |
Nelson’s syndrome | Often large: >10 mm | Post-adrenalectomy, pigmentation, sometimes local symptoms |
Non-functioning tumours | Usually large: >10 mm | Visual field defects; hypopituitarism (microadenomas may be incidental finding) |
Craniopharyngioma | Often very large and cystic (skull X-ray abnormal in >50%; calcification common) | Headaches, visual field defects, growth failure (50% occur below age 20; about 15% arise from within sella) |
Investigations (of a possible or proven mass)
Is there a tumour?
 the visual pathways, with field defects and visual loss (most common)
the visual pathways, with field defects and visual loss (most common)
 the cavernous sinus, with III, IV and VI cranial nerve lesions
the cavernous sinus, with III, IV and VI cranial nerve lesions
 bony structures and the meninges surrounding the fossa, causing headache
bony structures and the meninges surrounding the fossa, causing headache

 the ventricles, causing interruption of cerebrospinal fluid (CSF) flow leading to hydrocephalus
the ventricles, causing interruption of cerebrospinal fluid (CSF) flow leading to hydrocephalus

Investigations
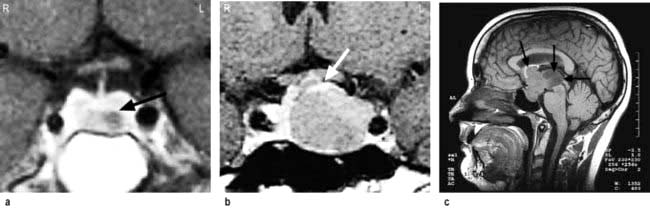
 MRI of the pituitary. MRI is superior to CT scanning (Fig. 19.8) and will readily show any significant pituitary mass. Small lesions within the pituitary fossa on MRI consistent with small pituitary microadenomas are very common (10% of normal individuals in some studies). Such small lesions are sometimes detected during MRI scanning of the head for other reasons – so-called ‘pituitary incidentalomas’.
MRI of the pituitary. MRI is superior to CT scanning (Fig. 19.8) and will readily show any significant pituitary mass. Small lesions within the pituitary fossa on MRI consistent with small pituitary microadenomas are very common (10% of normal individuals in some studies). Such small lesions are sometimes detected during MRI scanning of the head for other reasons – so-called ‘pituitary incidentalomas’.
 Visual fields. These should be plotted formally by automated computer perimetry or Goldmann perimetry, but clinical assessment by confrontation using a small red pin as target is also sensitive and valuable. Common defects are upper temporal quadrantanopia and bitemporal hemianopia (see p. 1073).
Visual fields. These should be plotted formally by automated computer perimetry or Goldmann perimetry, but clinical assessment by confrontation using a small red pin as target is also sensitive and valuable. Common defects are upper temporal quadrantanopia and bitemporal hemianopia (see p. 1073).
Is there a hormonal excess?
 Prolactin excess (prolactinoma or hyperprolactinaemia): histologically, prolactinomas are ‘chromophobe’ adenomas (a description of their appearance on classical histological staining)
Prolactin excess (prolactinoma or hyperprolactinaemia): histologically, prolactinomas are ‘chromophobe’ adenomas (a description of their appearance on classical histological staining)
 GH excess (acromegaly or gigantism): somatotroph adenomas, usually ‘acidophil’, and sometimes due to specific G-protein mutations (see p. 945)
GH excess (acromegaly or gigantism): somatotroph adenomas, usually ‘acidophil’, and sometimes due to specific G-protein mutations (see p. 945)

The clinical features of acromegaly, Cushing’s disease or hyperprolactinaemia are usually (but not always) obvious, and are discussed on pages 953, and 957. Hyperprolactinaemia may be clinically ‘silent’. Tumours producing LH, FSH or TSH are well described but very rare.
Is there a deficiency of any hormone?
Clinical examination may give clues; thus, short stature in a child with a pituitary tumour is likely to be due to GH deficiency. A slow, lethargic adult with pale skin is likely to be deficient in TSH and/or ACTH. Milder deficiencies may not be obvious, and require specific testing (see Table 19.7).
Treatment
Treatment depends on the type and size of tumour (Table 19.5). In general, therapy has three aims:
Table 19.5 Comparisons of primary treatments for pituitary tumours
| Treatment method | Advantages | Disadvantages |
|---|---|---|
Surgical |
|
|
Trans-sphenoidal adenomectomy or hypophysectomy | Relatively minor procedure Potentially curative for microadenomas and smaller macroadenomas | Some extrasellar extensions may not be accessible |
Transcranial (usually transfrontal) | Good access to suprasellar region | Major procedure; danger of frontal lobe damage |
Radiotherapy |
|
|
External (40–50 Gy) | Non–invasive | Slow action, often over many years |
Stereotactic | Precise administration of high dose to lesion | Long-term follow-up data limited |
Medical |
|
|
Dopamine agonist therapy (e.g. bromocriptine, cabergoline) | Non-invasive; reversible | Usually not curative; significant side-effects in minority |
Somatostatin analogue therapy (octreotide, lanreotide) | Non-invasive; reversible | Usually not curative; gallstones; expensive |
Growth hormone receptor antagonist (pegvisomant) | Highly selective | Usually not curative; very expensive |
Removal/control of tumour
 Surgery via the trans-sphenoidal route is usually the treatment of choice. Very large tumours are occasionally removed via the open transcranial (usually transfrontal) route.
Surgery via the trans-sphenoidal route is usually the treatment of choice. Very large tumours are occasionally removed via the open transcranial (usually transfrontal) route.

 Medical therapy with somatostatin analogues and/or dopamine agonists sometimes causes shrinkage of specific types of tumour (see p. 954) and if successful can be used as primary therapy.
Medical therapy with somatostatin analogues and/or dopamine agonists sometimes causes shrinkage of specific types of tumour (see p. 954) and if successful can be used as primary therapy.
Reduction of excess hormone secretion
Reduction is usually obtained by surgical removal but sometimes by medical treatment. Useful control can be achieved with dopamine agonists for prolactinomas or somatostatin analogues for acromegaly, but ACTH secretion usually cannot be controlled by medical means. Growth hormone antagonists are also available for acromegaly (p. 955).
Replacement of hormone deficiencies
Replacement of hormone deficiencies, i.e. hypopituitarism, is discussed below (see Table 19.8).
Table 19.8 Replacement therapy for hypopituitarism
| Axis | Usual replacement therapies |
|---|---|
Adrenal | Hydrocortisone 15–40 mg daily (starting dose 10 mg on rising/5 mg lunchtime/5 mg evening) |
(Normally no need for mineralocorticoid replacement) | |
Thyroid | Levothyroxine 100–150 µg daily |
Gonadal |
|
Male | Testosterone intramuscularly, orally, transdermally or implant |
Female | Cyclical oestrogen/progestogen orally or as patch |
Fertility | HCG plus FSH (purified or recombinant) or pulsatile GnRH to produce testicular development, spermatogenesis or ovulation |
Growth | Recombinant human GH used routinely to achieve normal growth in children |
Also advocated for replacement therapy in adults where GH has effects on muscle mass and wellbeing | |
Thirst | Desmopressin 10–20 µg one to three times daily by nasal spray or orally 100–200 µg three times daily |
Carbamazepine, thiazides and chlorpropamide are very occasionally used in mild diabetes insipidus | |
Breast (prolactin inhibition) | Dopamine agonist (e.g. cabergoline, 500 µg weekly) |
Differential diagnosis of pituitary or hypothalamic masses
Other tumours
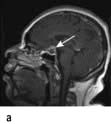
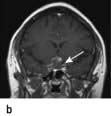
 Craniopharyngioma (1–2%), a usually cystic hypothalamic tumour, often calcified, arising from Rathke’s pouch, often mimics an intrinsic pituitary lesion. It is the most common pituitary tumour in children but may present at any age.
Craniopharyngioma (1–2%), a usually cystic hypothalamic tumour, often calcified, arising from Rathke’s pouch, often mimics an intrinsic pituitary lesion. It is the most common pituitary tumour in children but may present at any age.
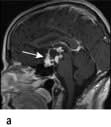
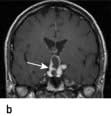
Craniopharyngioma: a partially cystic pituitary and suprasellar mass. (a) Sagittal, (b) coronal MRI.

Hypopituitarism
Causes
Disorders causing hypopituitarism are listed in Table 19.6. Pituitary and hypothalamic tumours, and surgical or radiotherapy treatment, are the most common.
Table 19.6 Causes of hypopituitarism
Congenital | Traumatic |
Isolated deficiency of pituitary hormones (e.g. Kallmann’s syndrome) | Skull fracture through base |
Infiltrations | |
Infective | Sarcoidosis |
Basal meningitis (e.g. tuberculosis) | |
Vascular | |
Pituitary apoplexy | |
Others | |
Radiation damage | |
Immunological | ‘Functional’ |
Autoimmune (lymphocytic) hypophysitis | Anorexia nervosa |
Neoplastic | |
Pituitary or hypothalamic tumours | |
Craniopharyngioma | |
Meningiomas | |
Gliomas | |
Pinealoma | |
Secondary deposits, especially breast | |
Lymphoma |
Clinical features
 Secondary hypothyroidism and adrenal failure both lead to tiredness and general malaise.
Secondary hypothyroidism and adrenal failure both lead to tiredness and general malaise.
 Hypothyroidism causes weight gain, slowness of thought and action, dry skin and cold intolerance.
Hypothyroidism causes weight gain, slowness of thought and action, dry skin and cold intolerance.


 Hyperprolactinaemia may cause galactorrhoea and hypogonadism.
Hyperprolactinaemia may cause galactorrhoea and hypogonadism.
 GH deficiency causes growth failure in children and impaired wellbeing in some adults.
GH deficiency causes growth failure in children and impaired wellbeing in some adults.


Kallmann’s syndrome. This syndrome is isolated gonadotrophin (GnRH) deficiency (p. 976).This syndrome arises due to mutations in the KAL1 gene which is located on the short (p) arm of the X chromosome. Kallmann’s is classically characterized by anosmia because the KAL1 gene provides instructions to make anosmin, which has a role in development of both the olfactory system as well as migration of GnRH secreting neurones.
Investigations
Tests range from the simple basal levels (e.g. free T4 for the thyroid axis), to stimulatory tests for the pituitary, and tests of feedback for the hypothalamus (Table 19.7). Assessment of the hypothalamic-pituitary-adrenal axis is complex: basal 09:00 hours cortisol levels above 400 nmol/L usually indicate an adequate reserve, while levels below 100 nmol/L predict an inadequate stress response. In many cases basal levels are equivocal and a dynamic test is essential: the insulin tolerance test (Box 19.2) is widely regarded as the ‘gold standard’ but the short ACTH stimulation test (Box 19.1), though an indirect measure, is used by many as a routine test of hypothalamic-pituitary-adrenal status. Occasionally, the difference between ACTH deficiency and normal HPA axis can be subtle, and the assessment of adrenal reserve is best left to an experienced endocrinologist.
 Box 19.2
Box 19.2
Insulin tolerance test

Procedure
 Test explained to patient and consent obtained
Test explained to patient and consent obtained
 Should only be performed in experienced, specialist units
Should only be performed in experienced, specialist units

 Intravenous hydrocortisone and glucose available for emergency
Intravenous hydrocortisone and glucose available for emergency
 Overnight fast, begin at 08:00–09:00 hours
Overnight fast, begin at 08:00–09:00 hours
 Soluble insulin, 0.15 U/kg, i.v. at time 0
Soluble insulin, 0.15 U/kg, i.v. at time 0
 Glucose, cortisol and GH levels at 0, 30, 45, 60, 90, 120 min
Glucose, cortisol and GH levels at 0, 30, 45, 60, 90, 120 min



Treatment
 Steroid and thyroid hormones are essential for life. Both are given as oral replacement drugs, as in primary thyroid and adrenal deficiency, aiming to restore the patient to clinical and biochemical normality (Table 19.8) and levels are monitored by routine hormone assays. Note: Thyroid replacement should not commence until normal glucocorticoid function has been demonstrated or replacement steroid therapy initiated, as an adrenal ‘crisis’ may otherwise be precipitated.
Steroid and thyroid hormones are essential for life. Both are given as oral replacement drugs, as in primary thyroid and adrenal deficiency, aiming to restore the patient to clinical and biochemical normality (Table 19.8) and levels are monitored by routine hormone assays. Note: Thyroid replacement should not commence until normal glucocorticoid function has been demonstrated or replacement steroid therapy initiated, as an adrenal ‘crisis’ may otherwise be precipitated.




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


