Chapter 36 Drugs to Control Pain
| Abbreviations | |
|---|---|
| CNS | Central nervous system |
| COX | Cyclooxygenase |
| GI | Gastrointestinal |
| 5-HT | Serotonin |
| IV | Intravenous |
| MAO | Monoamine oxidase |
| NAPQI | N-acetyl-p-benzoquinoneimine |
| NE | Norepinephrine |
| NSAID | Nonsteroidal antiinflammatory drug |
| PG | Prostaglandin |
| PGI2 | Prostacyclin |
| TX | Thromboxane |
Therapeutic Overview
The third group of analgesics includes compounds that do not relieve pain from tissue damage (i.e., nociceptive pain) but can provide relief in specific pain syndromes such as neuropathic pain, gout, migraine headache, and fibromyalgia. Neuropathic pain results from changes in sensory neurons that render them hyperactive, even in the absence of nociceptive stimuli. It is often a chronic condition that is impervious to standard analgesic drugs. However, neuropathic pain is ameliorated by tricyclic antidepressants and compounds used to treat seizure disorders, drugs that are not thought of as primary analgesic agents.
| Therapeutic Overview |
|---|
| Opioids |
| Relief of most types of moderate to severe visceral or somatic pain |
| Symptomatic treatment of acute diarrhea |
| Cough suppression |
| Treatment of opiate addiction and alcoholism |
| Anesthetic adjunct |
| Overdose can be reversed by opioid receptor antagonists |
| NSAIDs and Acetaminophen |
| Relief of mild to moderate somatic pain including headache, toothache, myalgia, and arthralgia |
| Reduce fever |
| Prophylaxis of myocardial infarction and stroke |
| NSAIDs only |
| Relief in inflammatory disorders including rheumatoid arthritis, osteoarthritis, gout, and ankylosing spondylitis |
Mechanisms of Action
Sensations of pain are modulated by both ascending and descending pathways in the central nervous system (CNS). Noxious or nociceptive stimuli activate highly developed endings on primary afferent neurons, termed nociceptors (pain receptors). These stimuli give rise to action potentials that are transmitted along afferent neurons into the dorsal horn of the spinal cord. A-delta (Aδ) fibers are small, myelinated, rapidly conducting afferent neurons that terminate in lamina I of the spinal cord. They have a relatively high threshold for activation by mechanical and thermal stimuli and mediate sharp and localized pain, often termed somatic pain. C-fibers are even smaller unmyelinated afferent neurons and hence are slower conducting. They are polymodal and are activated by mechanical, thermal, or chemical stimuli. They terminate in lamina II of the spinal cord (substantia gelatinosa) and mediate dull, diffuse, aching, or burning pain sometimes called visceral pain (see Chapter 13, Fig. 13-5). Aδ and C-fibers release excitatory amino acids in the dorsal horn; C-fibers, which are stimulated by bradykinin and prostaglandins (PGs) released from local damaged cells, release substance P and other neuropeptides. These neurotransmitters activate secondary neurons that form the ascending spinothalamic pathway, which projects to supraspinal nuclei in the thalamus and then to the limbic system and cerebral cortex (Fig. 36-1, A).
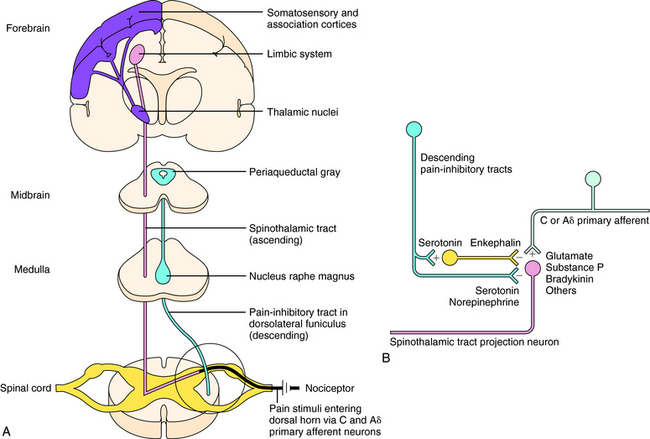
Descending pain-inhibitory systems originate in the periaqueductal gray region of the midbrain and from several nuclei of the rostroventral medulla oblongata and project downward to the dorsal horn. These descending systems release norepinephrine (NE), 5-HT, and other neurotransmitters and thereby inhibit the activity of the ascending pain pathways, either through direct synaptic contacts or indirectly by activating inhibitory interneurons. These pathways are illustrated in Figure 36-1, B.
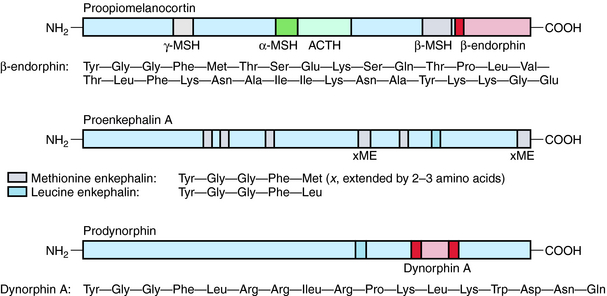
Three major families of opioid peptides have been identified: the enkephalins, endorphins, and dynorphins. They are derived from precursor molecules encoded by separate genes—proenkephalin, proopiomelanocortin, and prodynorphin, respectively. Although found primarily in the CNS, some opioid peptides, notably the enkephalins, also exist in peripheral tissues such as nerve plexuses of the gastrointestinal (GI) tract and the adrenal medulla (Table 36-1).
TABLE 36–1 Principal Endogenous Opioid Peptides
| Opioid Family | Precursor | Distribution |
|---|---|---|
| Enkephalins | Proenkephalin | Widely throughout the CNS, especially in interneurons, including those associated with pain pathways and emotional behavior; also found in some peripheral tissues |
| Endorphins | Proopiomelanocortin | β-Endorphin in hypothalamus, nucleus tractus solitarius, and anterior lobe of the pituitary where it is co-released with adrenocorticotrophin in response to stress |
| Dynorphins | Prodynorphin | Dynorphin A (1-17) in the magnocellular cells of the hypothalamus and posterior lobe of the pituitary gland where it co-localizes with vasopressin; shorter-chain dynorphins distributed widely in the CNS, some associated with pain pathways, especially in the spinal cord |
β-Endorphin and longer-chain dynorphins, such as dynorphin A (1-17), have a more limited distribution in the CNS and may not influence pain processing directly. Rather, they may have hormonal roles in responses to stress and fluid homeostasis, respectively. The structures of the three classical families of opioid peptides are shown in Figure 36-2.
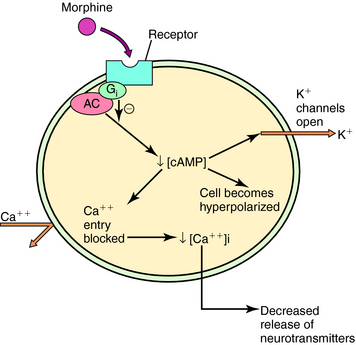
Three major opioid receptors have been identified by pharmacological means and molecular cloning and are designated μ, κ, and δ (Table 36-2). They are also referred to by either their International Union of Pharmacology nomenclature (OP3, OP2, and OP1, respectively) or their molecular biological nomenclature (MOP, KOP, and DOP, respectively). All three receptors belong to the superfamily of G-protein-coupled receptors with the characteristic seven transmembrane-spanning regions (see Chapter 1). Activation of these receptors decreases synthesis of cyclic adenosine monophosphate, increases K+ conductance, and decreases Ca++ conductance, effects illustrated in Figure 36-3. Because changes in K+ and Ca++ conductances inhibit neuronal activity, activation of any of the three opioid receptors usually results in decreased neuronal transmission.
TABLE 36–2 Opioid Receptors and their Ligands
| Receptor | Endogenous Ligand | Drug Ligands |
|---|---|---|
| μ Receptor (OP3/MOP) | Enkephalins | Morphine |
| β-Endorphin | Buprenorphine | |
| Endomorphins (?) | Methadone | |
| Meperidine | ||
| Fentanyl | ||
| κ Receptor (OP2/KOP) | Dynorphins | Butorphanol |
| Pentazocine | ||
| δ Receptor (OP1/DOP) | Enkephalins | None to date |
| β-Endorphin |
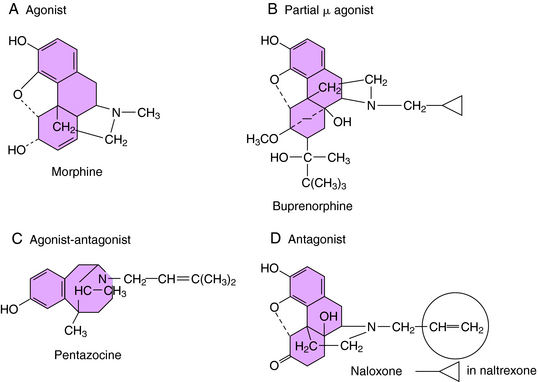
The selectivity of opioid receptors for endogenous and drug ligands is shown in Table 36-2. The anatomical distribution of opioid receptors is consistent with the actions of the opioids—that is, they are found prominently among structures of the ascending and descending pain-modulatory pathways. All clinically important effects of morphine and morphine-like drugs are mediated by μ receptors. Some of the effects of mixed-action opioids, including analgesia at the level of the spinal cord, sedation, and the dysphoria that occurs at high doses, are mediated by κ receptors. With the exception of the opioid antagonists, there are currently no therapeutic agents that interact with μ receptors in a clinically meaningful way. In cases in which opioids can be resolved into optical isomers, the levorotatory isomer usually has a considerably higher affinity for opioid receptors than does its dextrorotatory counterpart. The structures of morphine and representative agonist/antagonist compounds are shown in Figure 36-4.
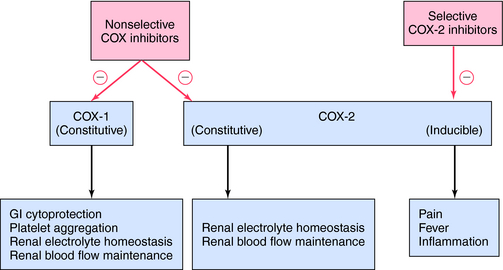
The mechanism of action, all of the therapeutic effects, and many of the side effects of the NSAIDs are due to inhibition of cyclooxygenase (COX), an enzyme involved in the metabolism of the eicosanoids. The eicosanoids are derivatives of arachidonic acid and include the leukotrienes synthesized by the action of 5-lipoxygenases, and the PGs and thromboxanes (TXs) synthesized by the action of the COXs (see Chapter 15, Fig. 15-1). Two distinct COX enzymes have been identified. COX-1 is constitutively expressed and is involved in “housekeeping tasks” in cells. COX-2 occurs constitutively in some tissues but is largely inducible, and induction results in a marked increase in the rate of synthesis and release of COX products, particularly the PGs. Aspirin acetylates both COX enzymes, inhibiting their activity irreversibly, whereas other nonselective NSAIDs inhibit the COX enzymes reversibly. The COX-2 inhibitors are 8- to 35-fold more selective for COX-2 relative to COX-1 and inhibit COX-2 irreversibly in a time-dependent manner. The functional consequences of inhibition of COX-2 relative to COX-1 are depicted in Figure 36-5. Structures of aspirin, acetaminophen, and the COX-2 inhibitor celecoxib are shown in Figure 36-6.
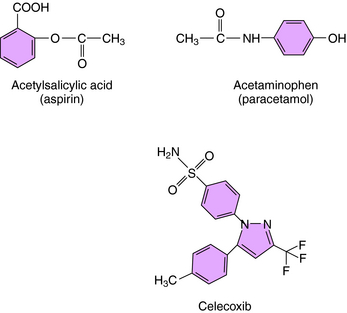
Drugs for Specific Pain Syndromes
Neuropathic pain is the result of injury to peripheral sensory nerves and is different from the nociceptive pain caused by tissue damage, in which sensory nerves are activated by chemical mediators of pain. There are many causes of nerve injury, including physical trauma, metabolic and autoimmune disorders, viral infection, chemotoxicity, and chronic inflammation. Nearly half of all diabetic patients experience peripheral neuropathies eventually. Neuropathic pain states often are associated with hyperalgesia (increased sensitivity to normally painful stimuli) and allodynia (pain caused by stimuli that are not normally painful; e.g., touch).
In neuropathic pain, primary afferent neurons are hyperactive, discharging spontaneously (in the absence of an identifiable noxious stimulus), and there is a cascade of changes to neurons in dorsal root ganglia and in the dorsal horn of the spinal cord. These changes present multiple targets for pharmacological intervention, among which are increases in the expression and activity of Na+ channels. Several drugs introduced to treat seizure disorders have been shown to be effective in the treatment of neuropathic pain. These agents include carbamazepine and lamotrigine, which inhibit voltage-dependent Na+ channels, and pregabalin, which binds to an auxiliary subunit of voltage-gated Ca++ channels to decrease the release of several neurotransmitters (see Chapter 34). The tricyclic antidepressants (see Chapter 30) have also been shown to be effective for this condition.
Drugs from several pharmacological classes are used to treat or prevent gout. The NSAIDs and the corticosteroids (see Chapter 39) attenuate inflammatory responses to urate crystals and the associated pain. Colchicine also reduces the inflammatory response, but through a different mechanism. Colchicine binds to tubulin in leukocytes, causing microtubules to disaggregate. This affects the structure of the cells, inhibiting their migration into the inflamed area and reducing phagocytic activity.
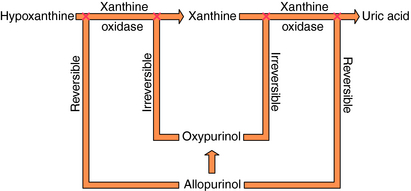
Specific drugs are used to correct the underlying hyperuricemia in gout. Allopurinol, a structural analog of the purine hypoxanthine, inhibits the enzyme xanthine oxidase (Fig. 36-7), blocking the metabolism of hypoxanthine and xanthine to uric acid and lowering blood urate concentrations. Normally, approximately 90% of filtered urate is resorbed and only 10% is excreted. The uricosuric agents probenecid and sulfinpyrazone increase urate excretion by competing with uric acid for the renal tubular acid transporter so less urate is resorbed.
The NSAIDs also bring relief from migraine episodes in many patients. They are presumed to attenuate the neurogenically induced inflammatory response through inhibition of COX-2. Other drugs are also used as preventive therapy, including tricyclic antidepressants, especially amitriptyline (see Chapter 30), and the β adrenergic receptor blockers propranolol and timolol (see Chapter 11).



