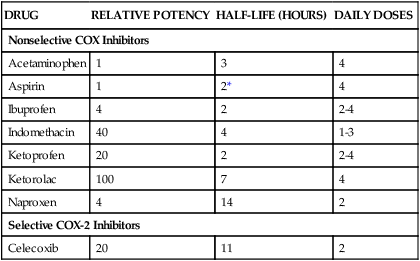
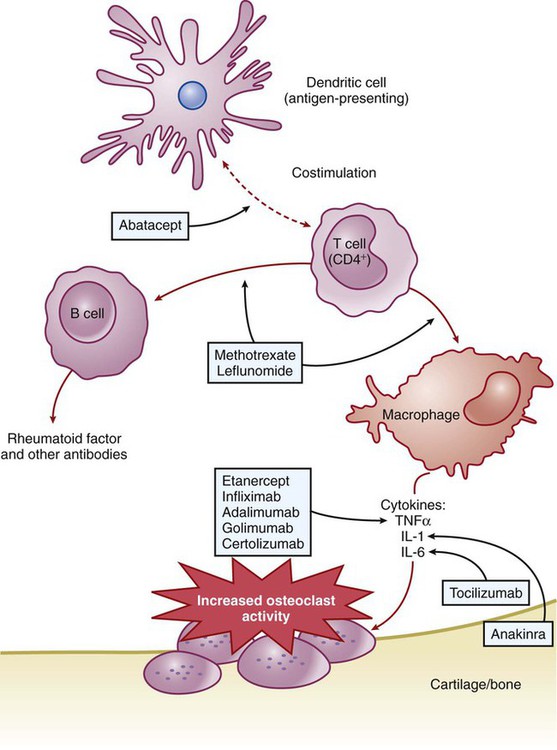
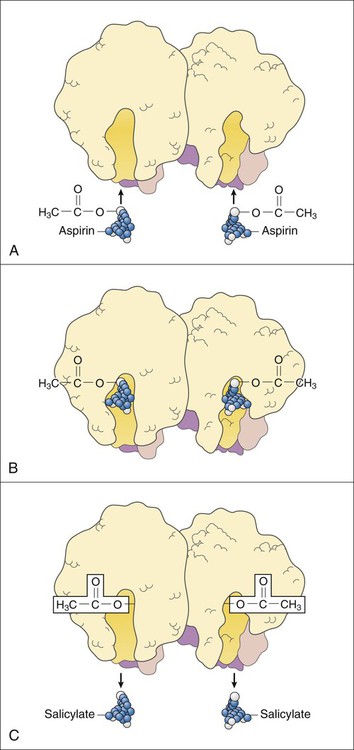
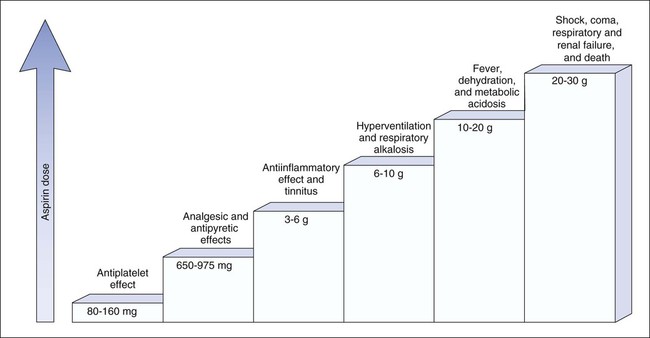
RA is an autoimmune disorder of unknown cause. The hallmark symptom of RA is joint inflammation, and most patients with RA experience a chronic, fluctuating course of disease that, despite therapeutic measures, can result in progressive joint destruction, deformity, disability, and premature death. RA affects 2% to 3% of the U.S. population, making it the most common systemic inflammatory disease (Box 30-1). It is three times more common in women than in men. RA is characterized by symmetrical joint inflammation that most frequently affects the small joints of the hands, wrists, and feet, but also the joints of the ankles, elbows, hips, knees, and shoulders. Cardiopulmonary, neurologic, and ocular inflammation are also often found in patients with RA, and many patients develop rheumatoid nodules on the extensor surfaces of the elbows, forearms, and hands. In addition, many patients have extraarticular manifestations, such as vasculitis, lymphadenopathy, and splenomegaly. As shown in Figure 30-1, RA is triggered by autoimmune mechanisms that lead to the destruction of synovial tissue and other connective tissue. Both humoral and cellular immune mechanisms are involved in the pathogenesis of the disease. These mechanisms include the cytokine-mediated activation of T and B lymphocytes and the recruitment and activation of macrophages. The inflammatory leukocytes then release a variety of prostaglandins, cytotoxic compounds, and free radicals that cause joint inflammation and destruction. Patients with RA show elevated levels of immunoglobulin G–rheumatoid factor (IgG-RF) complexes; extracorporeal filtering of these complexes using immunoabsorption apheresis has helped some patients. In patients with RA, NSAIDs are used to relieve pain and inflammation, and DMARDs are used to suppress the underlying disease process and slow the progression of joint destruction. The sites of action of selected antirheumatic drugs are depicted in Figure 30-1 and discussed later. Nonpharmacologic measures for treating OA include joint protection and splinting, physiotherapy, orthotic prostheses to support the feet, and joint replacement surgery. Pharmacologic measures include NSAIDs, local glucocorticoid injections, and experimental chondroprotective drugs (e.g., chondroitin sulfate and glucosamine). Recently, sodium hyaluronate (SUPARTZ) was approved for intraarticular injection as a type of joint fluid replacement in the treatment of OA. It is a sterile, viscoelastic solution prepared from chicken combs (the fleshy growths on top of chicken heads). The antidepressant duloxetine (see Chapter 22) is also indicated for the management of persistent musculoskeletal pain caused by chronic OA and chronic low back pain. The NSAIDs make up a large family of weak acidic drugs whose pharmacologic effects result primarily from the inhibition of cyclooxygenase (COX), an enzyme that catalyzes the first step in the synthesis of prostaglandins from arachidonic acid and other precursor fatty acids (see Chapter 26). COX is a microsomal enzyme, existing as a dimer (two molecules linked to form a functional unit) in the lumen and membrane of the endoplasmic reticulum. NSAIDs decrease COX activity primarily by competitive inhibition; however, aspirin forms a covalent, irreversible inhibition of COX (Fig. 30-2). The net effect of NSAID administration is a decrease in the production of prostaglandins and other autacoids. Among the nonselective COX inhibitors are many well-known NSAIDs that are available without a prescription, including aspirin, ibuprofen, ketoprofen, and naproxen. Acetaminophen is a weak antiinflammatory agent, but it is also included in this class of drugs because it exerts analgesic and antipyretic effects via inhibition of COX. As shown in Table 30-1, NSAIDs vary greatly in potency and half-life, but most of them are administered two to four times a day with food. TABLE 30-1 Properties of Nonsteroidal Antiinflammatory Drugs *For aspirin, the value shown is the half-life of the active metabolite, salicylic acid. The salicylates are usually administered orally, but formulations are also available for topical and rectal administration. The oral dosage of aspirin that is needed to inhibit platelet aggregation is somewhat lower than the oral dosage needed to obtain analgesic and antipyretic effects, and it is much lower than the oral dosage needed to relieve inflammation caused by arthritic and other inflammatory disorders. Figure 30-3 shows the relationship between the dosage of aspirin and the pharmacologic and toxic effects of the drug. Most of the salicylic acid formed from aspirin and other salicylate drugs is conjugated with glycine to form salicyluric acid. This substance is then excreted in the urine, along with about 10% of free salicylate and a similar amount of glucuronide conjugates. The rate of excretion of salicylate is affected by urine pH. For this reason, alkalinization of the urine by administration of sodium bicarbonate has been used to increase the ionization and elimination of salicylic acid in cases of drug overdose (see Chapter 2).
Drugs for Pain, Inflammation, and Arthritic Disorders
Rheumatoid Arthritis
Osteoarthritis
Nonsteroidal Antiinflammatory Drugs
Mechanism of Action
Specific Agents
Nonselective Cyclooxygenase Inhibitors
DRUG
RELATIVE POTENCY
HALF-LIFE (HOURS)
DAILY DOSES
Nonselective COX Inhibitors
Acetaminophen
1
3
4
Aspirin
1
2*
4
Ibuprofen
4
2
2-4
Indomethacin
40
4
1-3
Ketoprofen
20
2
2-4
Ketorolac
100
7
4
Naproxen
4
14
2
Selective COX-2 Inhibitors
Celecoxib
20
11
2
Aspirin and Other Salicylates
Pharmacologic Effects and Indications
Pharmacokinetics
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Drugs for Pain, Inflammation, and Arthritic Disorders
Only gold members can continue reading. Log In or Register to continue
