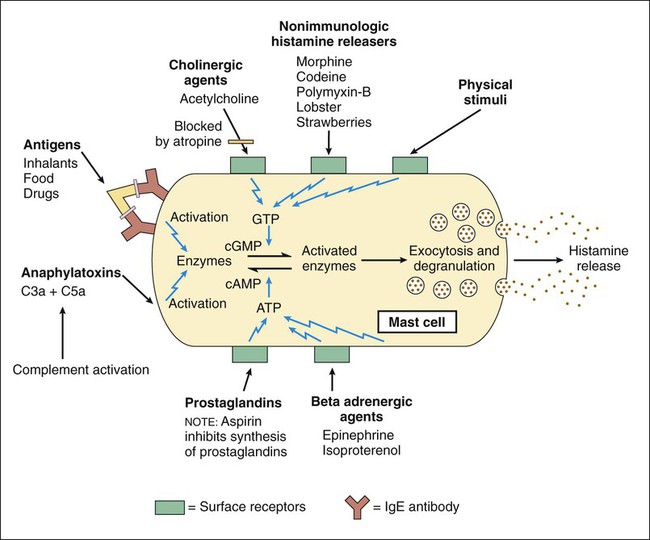
Autacoids (also spelled autocoids) are substances produced by neural and nonneural tissues throughout the body that act locally to modulate the activity of smooth muscles, nerves, glands, platelets, and other tissues (Table 26-1). Several autacoids also serve as neurotransmitters in the central nervous system (CNS) or enteric nervous system. TABLE 26-1 NVSM, Nonvascular smooth muscle; VSM, vascular smooth muscle. Histamine is a biogenic amine produced primarily by mast cells and basophils, which are particularly abundant in the skin, gastrointestinal tract, and respiratory tract. Histamine is also produced by paracrine cells in the gastric fundus, where it stimulates acid secretion by parietal cells. Histamine also functions as a neurotransmitter in the CNS (see Chapter 18). Histamine is formed when the amino acid histidine is decarboxylated in a reaction catalyzed by the enzyme L–histidine decarboxylase. Histamine is stored in granules (vesicles) in mast cells and basophils until it is released. It is released from mast cells when membrane-bound immunoglobulin E (IgE) interacts with an IgE antigen to cause mast cell degranulation. This process can be blocked by cromolyn sodium and related respiratory drugs, as described in Chapter 27. A number of other stimuli can also cause the release of histamine from mast cells (Fig. 26-1). Stimuli that increase cyclic guanosine monophosphate increase histamine release, whereas those that increase cyclic adenosine monophosphate oppose this action. Antihistamines, or histamine receptor antagonists, have been categorized on the basis of their receptor selectivity as H1 receptor antagonists or H2 receptor antagonists. Chapter 28 outlines the properties of H2 receptor antagonists, which are used primarily to treat peptic ulcer disease. There are presently no approved H3 receptor agents, although clinical trials are underway. The H1 antihistamines are all equally effective in treating allergies, but they differ markedly in their sedative, antiemetic, and anticholinergic properties (Table 26-2). The second-generation antihistamines cause little or no sedation, so they are often preferred for the treatment of allergies. Antihistamines are usually more effective when administered before exposure to an allergen than afterward. Hence persons with seasonal allergies, such as allergic rhinitis (see Chapter 27), should take them on a regular basis throughout the allergy season. TABLE 26-2 Pharmacologic Properties of Selected Histamine H1 Receptor Antagonists Meclizine, diphenhydramine, hydroxyzine, and promethazine have higher antiemetic activity than other antihistamines. Meclizine is less sedating than diphenhydramine, hydroxyzine, and promethazine, so it is frequently used to prevent motion sickness or treat vertigo. Dimenhydrinate is a mixture of diphenhydramine and 8-chlorotheophylline and is also used for these purposes. Promethazine suppositories are often used to relieve nausea and vomiting associated with various conditions (see Chapter 28).
Autacoid Drugs
Overview
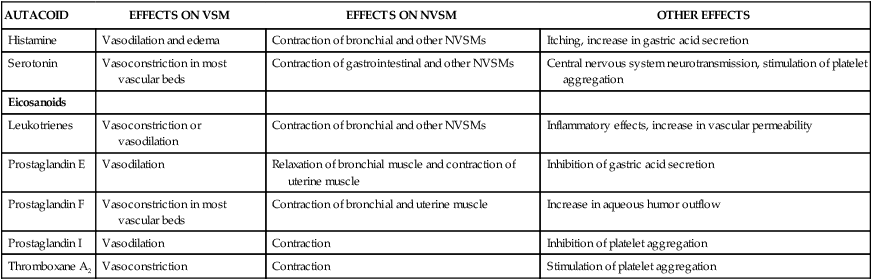
AUTACOID
EFFECTS ON VSM
EFFECTS ON NVSM
OTHER EFFECTS
Histamine
Vasodilation and edema
Contraction of bronchial and other NVSMs
Itching, increase in gastric acid secretion
Serotonin
Vasoconstriction in most vascular beds
Contraction of gastrointestinal and other NVSMs
Central nervous system neurotransmission, stimulation of platelet aggregation
Eicosanoids
Leukotrienes
Vasoconstriction or vasodilation
Contraction of bronchial and other NVSMs
Inflammatory effects, increase in vascular permeability
Prostaglandin E
Vasodilation
Relaxation of bronchial muscle and contraction of uterine muscle
Inhibition of gastric acid secretion
Prostaglandin F
Vasoconstriction in most vascular beds
Contraction of bronchial and uterine muscle
Increase in aqueous humor outflow
Prostaglandin I
Vasodilation
Contraction
Inhibition of platelet aggregation
Thromboxane A2
Vasoconstriction
Contraction
Stimulation of platelet aggregation
Histamine and Related Drugs
Histamine Biosynthesis and Release
Antihistamine Drugs
Histamine H1 Receptor Antagonists
Classification
Pharmacologic Effects and Indications
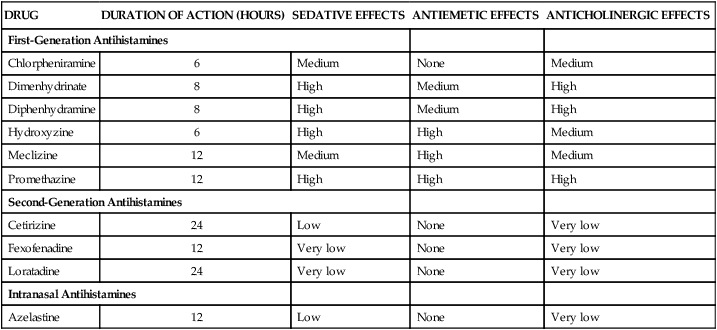
DRUG
DURATION OF ACTION (HOURS)
SEDATIVE EFFECTS
ANTIEMETIC EFFECTS
ANTICHOLINERGIC EFFECTS
First-Generation Antihistamines
Chlorpheniramine
6
Medium
None
Medium
Dimenhydrinate
8
High
Medium
High
Diphenhydramine
8
High
Medium
High
Hydroxyzine
6
High
High
Medium
Meclizine
12
Medium
High
Medium
Promethazine
12
High
High
High
Second-Generation Antihistamines
Cetirizine
24
Low
None
Very low
Fexofenadine
12
Very low
None
Very low
Loratadine
24
Very low
None
Very low
Intranasal Antihistamines
Azelastine
12
Low
None
Very low
First-Generation Antihistamines
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Autacoid Drugs
Only gold members can continue reading. Log In or Register to continue
