http://evolve.elsevier.com/McCuistion/pharmacology
Myasthenia gravis (MG) is an acquired autoimmune disease that impairs the transmission of messages at the neuromuscular junction, resulting in fluctuating muscle weakness that increases with muscle use. MG causes fatigue and muscular weakness of the respiratory system, facial muscles, and extremities. Due to cranial nerve involvement, ptosis (drooping eyelid) and difficulty in chewing and swallowing occur. Respiratory arrest may result from respiratory muscle paralysis. The symptoms of MG are caused by autoimmune destruction of acetylcholine (ACh) sites and a resultant decrease in neuromuscular transmission.
Multiple sclerosis (MS) is a neuromuscular autoimmune disorder that attacks the myelin sheath of nerve fibers, causing lesions known as plaques. Although there are no definitive diagnostic tests, the sclerotic plaques are usually detected and measured by magnetic resonance imaging (MRI). Pharmacologic treatment is necessary to control the symptoms of this disorder.
Muscle spasms have various causes, including injury or motor neuron disorders that are associated with conditions such as MS, MG, cerebral palsy, spinal cord injuries (paraplegia [paralysis of the legs]), cerebrovascular accident (CVA [stroke]), or hemiplegia (paralysis of one side of the body). Spasticity of muscles can be reduced with the use of skeletal muscle relaxants.
Myasthenia Gravis
MG is a chronic autoimmune neuromuscular disease that affects approximately 20 in 100,000 persons. It is estimated that 60,000 Americans are affected. MG can occur in people of any ethnicity and sex; however, MG peaks in women around the childbearing years, whereas the peak onset in men is between 50 and 70 years. MG can also occur in people outside of this age range. Although it is not a genetic disorder, a familial tendency may be apparent.
Pathophysiology
MG results from a lack of acetylcholine receptor (AChR) sites. This autoimmune disorder involves an antibody response against an alpha subunit of the AChR site at the neuromuscular junction. Antibodies attack the AChR sites, obstructing the binding of ACh and eventually destroying the receptor sites. When AChR sites are reduced, ACh molecules are prevented from binding to receptors and stimulating normal neuromuscular transmission. The result is ineffective muscle contraction and muscle weakness. About 90% of patients with MG have anti-ACh antibodies that can be detected through serum testing.
The thymus gland is involved in systemic immunity that is active during infancy and early childhood, but the gland normally shrinks during adulthood. Approximately 60% of MG patients have thymic hyperplasia. It has been suggested in some cases that if the thymus gland is removed during the early onset of MG, clinical symptoms are greatly decreased. Thymectomy has been an option for patients younger than 50 years.
MG is characterized primarily by weakness and fatigue of the skeletal muscles. In 90% of cases, eyelid or extraocular muscles are involved. The patient may experience ptosis and diplopia (double vision). Other characteristics of MG include dysphagia (difficulty chewing and swallowing), dysarthria (slurred speech), and respiratory muscle weakness.
The group of drugs used to control MG are the acetylcholinesterase (AChE) inhibitors. They inhibit the action of the enzyme AChE. As a result of this action, more ACh is available to activate the cholinergic receptors and promote muscle contraction. The AChE inhibitors are classified as parasympathomimetics.
When muscular weakness in the patient with MG becomes generalized, myasthenic crisis may occur. This complication is a severe, generalized muscle weakness that may involve the muscles of respiration, such as the diaphragm and intercostal muscles. Triggers of myasthenic crisis include inadequate dosing of AChE inhibitors, infection, emotional stress, menses, pregnancy, surgery, trauma, hypokalemia, temperature extremes, and alcohol intake. Myasthenic crisis can also occur 3 to 4 hours after taking certain medications (e.g., aminoglycoside and fluoroquinolone antibiotics, calcium channel blockers, phenytoin, and psychotropics; Box 21.1). If muscle weakness remains untreated, death could result from paralysis of the respiratory muscles. Neostigmine, a fast-acting AChE inhibitor, can relieve myasthenic crisis.
Overdosing with AChE inhibitors may cause another complication of MG called cholinergic crisis, which is an acute exacerbation of symptoms. A cholinergic crisis usually occurs within 30 to 60 minutes after taking anticholinergic drugs. This complication is due to continuous depolarization of postsynaptic membranes that creates a neuromuscular blockade. The patient with cholinergic crisis often has severe muscle weakness that can lead to respiratory paralysis and arrest. Accompanying symptoms include miosis (abnormal pupil constriction), pallor, sweating, vertigo, excess salivation, nausea, vomiting, abdominal cramping, diarrhea, bradycardia, and fasciculations (involuntary muscle twitching).
Acetylcholinesterase Inhibitors
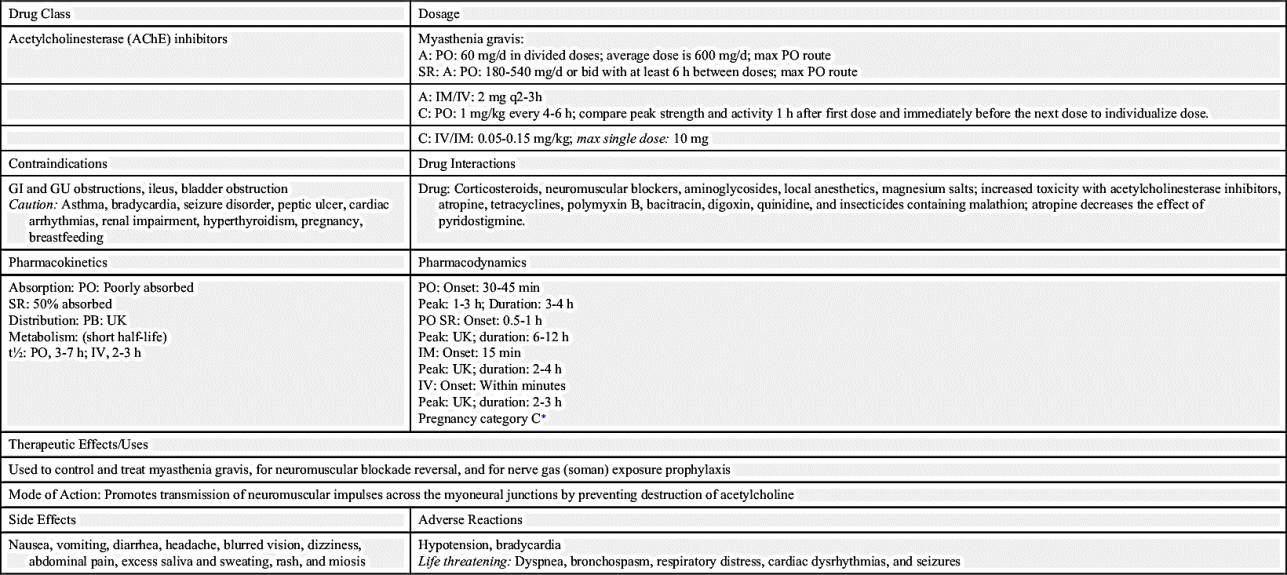
The first drug used to manage MG is neostigmine. It is a short-acting acetylcholinesterase inhibitor with a half-life of 0.5 to 1 hour. The drug must be given on time to prevent muscle weakness. The AChE inhibitor pyridostigmine has an intermediate action and is given every 4 to 6 hours (Table 21.1). Pyridostigmine is presented in Prototype Drug Chart 21.1.
Pharmacokinetics
Pyridostigmine is poorly absorbed from the gastrointestinal (GI) tract. Half of the sustained-release capsule is absorbed readily, but the balance is poorly absorbed. The half-life of oral pyridostigmine is 3 to 7 hours, and it is 2 to 3 hours for intravenous (IV) administration. Because of its short half-life, pyridostigmine must be administered several times a day. The drug is metabolized by the liver and is excreted in the urine.
Pharmacodynamics
Pyridostigmine increases muscle strength in patients with muscular weakness resulting from MG. The onset of action of oral pyridostigmine is 30 to 45 minutes, the peak is 1 to 3 hours, and the duration is 3 to 4 hours. Overdosing of pyridostigmine can result in signs and symptoms of cholinergic crisis. This crisis requires emergency medical intervention because of respiratory muscle weakness.
Patients who do not respond to AChE inhibitors may require additional drug treatment such as with prednisone, plasma exchange, IV immune globulin, or immunosuppressive drugs. Prednisone is the drug of choice, but like other immunosuppressants, it reduces the presence of antibodies. Corticosteroids do not produce permanent remission, and the long-term side effects are significant.
The immunosuppressive agent azathioprine can be used in conjunction with a lower dose of prednisone. With azathioprine, the white blood cell (WBC) count and liver enzymes should be closely monitored to avoid leukopenia and hepatotoxicity.
Overdosing and underdosing of AChE inhibitors have similar symptoms: generalized muscle weakness, which can include the muscles of respiration, the diaphragm, and the intercostal muscles resulting in dyspnea (difficulty breathing), and dysphagia. Additional symptoms that may be present with overdosing are increased salivation (drooling), sweating, and bronchial secretions, along with miosis, bradycardia, and abdominal pain. All doses of AChE inhibitors should be administered on time because late administration of the drug could result in muscle weakness.
TABLE 21.1
Acetylcholinesterase Inhibitors for Myasthenia Gravis
| Drug | Route and Dosage | Uses and Considerations |
| Edrophonium | A: IV 2 mg over 15-30 s; if no response, administer 8 mg; if cholinergic reaction occurs, administer 0.4-0.5 mg IV atropine. A: IM: 10 mg; if cholinergic reaction occurs, retest after 30 min with 2 mg to rule out false negative. C: <34 kg: IV: 1 mg, repeat in 45 s; if no response, give 1 mg q30-45s until response is seen; max: 5 mg C: >34 kg: IV: 2 mg, repeat with 1 mg if no response; max: 10 mg | For diagnosing MG. Will distinguish between myasthenic and cholinergic crisis. Ptosis should be absent in 1-5 min. Duration: IV: 5-10 min IM: 5-30 min Onset: Ultra–short-acting drug IV: Rapid 30-60 s IM: Rapid 2-10 min PB: UK; t½: 1.2-2 h Pregnancy category C∗ |
| Neostigmine | (The oral drug was discontinued in the United States.) A: IM/subcut: 0.5 mg/mL; 1 mg/mL; max: 5 mg A/C: IV: 0.03-0.07 mg/kg/d over 1 min Note: All anticholinesterase drugs should be discontinued 8 h before administration of neostigmine. | For controlling MG. Dose should be individualized to the patient. Must be given on time to prevent myasthenic crisis. Overdose can cause cholinergic reaction: nausea, abdominal cramps, excessive salivation, and sweating. Note: Large parenteral doses should be accompanied with IV atropine to counteract the side effects. PB: 15%-25%; t½: IM: 50-90 min; IV: 24-113 min Pregnancy category C∗ |
| Pyridostigmine | See Prototype Drug Chart 21.1. |

Underdosing can result in myasthenic crisis, and overdosing can result in cholinergic crisis. Edrophonium is an ultra–short-acting AChE inhibitor that may be used to distinguish between myasthenic crisis and cholinergic crisis. These two crises have a similar major symptom: severe muscle weakness. After edrophonium is administered, if the symptoms are alleviated because of an increase in ACh, the cause is myasthenic crisis. However, if the muscle weakness becomes more severe, the cause is cholinergic crisis due to drug overdosing. Edrophonium may also be used to diagnose MG. Its ultrashort duration of 5 to 30 minutes increases muscle strength immediately. If ptosis is immediately corrected after administration of this drug, the diagnosis is most likely MG.
Side Effects and Adverse Reactions
Side effects and adverse reactions of AChE inhibitors include GI disturbances (nausea, vomiting, diarrhea, abdominal cramps), increased salivation and tearing, miosis (constricted pupil of the eye), blurred vision, bradycardia, and hypotension.
 Nursing Process: Patient-Centered Collaborative Care
Nursing Process: Patient-Centered Collaborative CareDrug Treatment for Myasthenia Gravis: Pyridostigmine
Assessment
• Obtain a drug history from the patient that includes all current medications.
• Observe the patient’s drug profile for possible drug interactions.
• Patients should avoid atropine, atropine-like drugs, and muscle relaxants.
• Record baseline vital signs.
• Assess patient for signs and symptoms of myasthenic crisis, such as muscle weakness with difficulty breathing and swallowing.
Nursing Diagnoses