If cardiac dilation is insufficient to maintain cardiac output, other factors come into play. As discussed later, these are not always beneficial.
Increased Sympathetic Tone
HF causes arterial pressure to fall. In response, the baroreceptor reflex increases sympathetic output to the heart, veins, and arterioles. At the same time, parasympathetic effects on the heart are reduced. The consequences of increased sympathetic tone are summarized here.
• Increased heart rate. Acceleration of heart rate increases cardiac output, thereby helping improve tissue perfusion. However, if heart rate increases too much, there will be insufficient time for complete ventricular filling, and cardiac output will fall.
• Increased contractility. Increased myocardial contractility has the obvious benefit of increasing cardiac output. The only detriment is an increase in cardiac oxygen demand.
• Increased venous tone. Elevation of venous tone increases venous pressure and thereby increases ventricular filling. Because of Starling’s mechanism, increased filling increases stroke volume. Unfortunately, if venous pressure is excessive, blood will back up behind the failing ventricles, thereby aggravating pulmonary and peripheral edema. Furthermore, excessive filling pressure can dilate the heart so much that stroke volume will begin to decline.
• Increased arteriolar tone. Elevation of arteriolar tone increases arterial pressure, thereby increasing perfusion of vital organs. Unfortunately, increased arterial pressure also means the heart must pump against greater resistance. Because cardiac reserve is minimal in HF, the heart may be unable to meet this challenge, and output may fall.
Water Retention and Increased Blood Volume
Mechanisms
Water retention results from two mechanisms. First, reduced cardiac output causes a reduction in renal blood flow, which in turn decreases glomerular filtration rate (GFR). As a result, urine production is decreased and water is retained.
Second, HF activates the RAAS. Activation occurs in response to reduced blood pressure and reduced renal blood flow. When activated, the RAAS promotes water retention by increasing circulating levels of aldosterone and angiotensin II. Aldosterone acts directly on the kidneys to promote retention of sodium and water. Angiotensin II causes constriction of renal blood vessels, which decreases renal blood flow and thereby further decreases urine production. In addition, angiotensin II causes constriction of systemic arterioles and veins and thereby increases venous and arterial pressure.
Consequences
As with other adaptive responses to HF, increased blood volume can be beneficial or harmful. Increased blood volume increases venous pressure and thereby increases venous return. As a result, ventricular filling and stroke volume are increased. The resultant increase in cardiac output can improve tissue perfusion. However, as noted, if venous pressure is too high, edema of the lungs and periphery may result. More important, if the increase in cardiac output is insufficient to maintain adequate kidney function, renal retention of water will progress unabated. The resultant accumulation of fluid will cause severe cardiac, pulmonary, and peripheral edema—and, ultimately, death.
Natriuretic Peptides
In response to stretching of the atria and dilation of the ventricles, the heart releases two natriuretic peptides: atrial natriuretic peptide (ANP) and B-natriuretic peptide (BNP). As discussed in Chapter 34, these hormones promote dilation of arterioles and veins and also promote loss of sodium and water through the kidneys. Hence they tend to counterbalance vasoconstriction caused by the SNS and angiotensin II, as well as retention of sodium and water caused by the RAAS. However, as HF progresses, the effects of ANP and BNP eventually become overwhelmed by the effects of the SNS and RAAS.
Levels of circulating BNP are an important index of cardiac status in HF patients and thus can be a predictor of long-term survival. High levels of BNP indicate poor cardiac health and can predict a lower chance of survival. Conversely, low levels of BNP indicate better cardiac health and can predict a higher chance of survival. This information can be helpful when assessing the hospitalized patient at discharge: the lower the BNP level, the greater the chances of long-term survival.
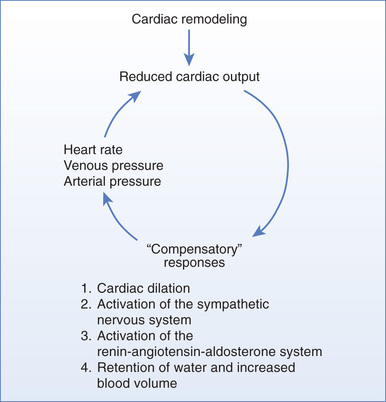
The Vicious Cycle of “Compensatory” Physiologic Responses
As discussed previously, reduced cardiac output leads to compensatory responses: (1) cardiac dilation, (2) activation of the SNS, (3) activation of the RAAS, and (4) retention of water and expansion of blood volume. Although these responses represent the body’s attempt to compensate for reduced cardiac output, they can actually make matters worse: excessive heart rate can reduce ventricular filling; excessive arterial pressure can lower cardiac output; and excessive venous pressure can cause pulmonary and peripheral edema. Thus, as depicted in Fig. 40.2, the “compensatory” responses can create a self-sustaining cycle of maladaptation that further impairs cardiac output and tissue perfusion. If cardiac output becomes too low to maintain sufficient production of urine, the resultant accumulation of water will eventually be fatal. The actual cause of death is complete cardiac failure secondary to excessive cardiac dilation and cardiac edema.
Signs and Symptoms of Heart Failure
The prominent signs and symptoms of HF are a direct consequence of the pathophysiology just described. Decreased tissue perfusion results in reduced exercise tolerance, fatigue, and shortness of breath; shortness of breath may also reflect pulmonary edema. Increased sympathetic tone produces tachycardia. Increased ventricular filling, reduced systolic ejection, and myocardial hypertrophy result in cardiomegaly (increased heart size). The combination of increased venous tone plus increased blood volume helps cause pulmonary edema, peripheral edema, hepatomegaly (increased liver size), and distention of the jugular veins. Weight gain results from fluid retention.
Classification of Heart Failure Severity
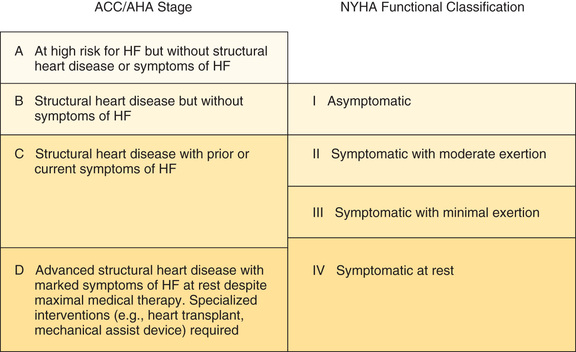
There are two major schemes for classifying HF severity. One scheme, established by the New York Heart Association (NYHA), classifies HF based on the functional limitations it causes. A newer scheme, proposed jointly by the American College of Cardiology (ACC) and the American Heart Association (AHA), is based on the observation that HF is a progressive disease that moves through stages of increasing severity.
The NYHA scheme, which has four classes, can be summarized as follows:
• Class I—no limitation of ordinary physical activity
• Class II—slight limitation of physical activity: normal activity produces fatigue, dyspnea, palpitations, or angina
• Class III—marked limitation of physical activity: even mild activity produces symptoms
The ACC/AHA scheme, which also has four stages, can be summarized as follows:
• Stage A—at high risk for HF but without structural heart disease or symptoms of HF
• Stage B—structural heart disease but without symptoms of HF
• Stage C—structural heart disease with prior or current symptoms of HF
The ACC/AHA scheme was unveiled in treatment guidelines issued in 2001. The newest version of that document—2013 ACCF/AHA Guideline for the Management of Heart Failure. Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines—is discussed later under “Management of Heart Failure.”
Please note that the ACC/AHA scheme is intended to complement the NYHA scheme, not replace it. The relationship between the two is shown in Fig. 40.3.
Overview of Drugs Used to Treat Heart Failure
For routine therapy, HF is treated with three types of drugs: (1) diuretics, (2) agents that inhibit the RAAS, and (3) beta blockers. Other agents (e.g., digoxin, dopamine, hydralazine) may be used as well.
Diuretics
Diuretics are first-line drugs for all patients with signs of volume overload or with a history of volume overload. By reducing blood volume, these drugs can decrease venous pressure, arterial pressure (afterload), pulmonary edema, peripheral edema, and cardiac dilation. However, excessive diuresis must be avoided: if blood volume drops too low, cardiac output and blood pressure may fall precipitously, thereby further compromising tissue perfusion. For the most part, benefits of diuretics are limited to symptom reduction. As a rule, these drugs do not prolong survival. The basic pharmacology of the diuretics is discussed in Chapter 35.
Thiazide Diuretics
The thiazide diuretics (e.g., hydrochlorothiazide) produce moderate diuresis. These oral agents are used for long-term therapy of HF when edema is not too great. Because thiazides are ineffective when GFR is low, these drugs cannot be used if cardiac output is greatly reduced. The principal adverse effect of the thiazides is hypokalemia, which increases the risk for digoxin-induced dysrhythmias (see later).
Loop Diuretics
The loop diuretics (e.g., furosemide) produce profound diuresis. In contrast to the thiazides, these drugs can promote fluid loss even when GFR is low. Therefore loop diuretics are preferred to thiazides when cardiac output is greatly reduced. Because they can mobilize large volumes of water, and because they work when GFR is low, loop diuretics are drugs of choice for patients with severe HF. Like the thiazides, these drugs can cause hypokalemia, thereby increasing the risk for digoxin toxicity. In addition, loop diuretics can cause severe hypotension secondary to excessive volume reduction.
Potassium-Sparing Diuretics
In contrast to the thiazides and loop diuretics, the potassium-sparing diuretics (e.g., spironolactone, triamterene) promote only scant diuresis. In patients with HF, these drugs are employed to counteract potassium loss caused by thiazide and loop diuretics, thereby lowering the risk for digoxin-induced dysrhythmias. Not surprisingly, the principal adverse effect of the potassium-sparing drugs is hyperkalemia. Because angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) also carry a risk for hyperkalemia, caution is needed if these drugs are combined with a potassium-sparing diuretic. Accordingly, when therapy with an ACE inhibitor or ARB is initiated, the potassium-sparing diuretic should be discontinued. It can be resumed later if needed.
One potassium-sparing diuretic—spironolactone—prolongs survival in patients with HF primarily by blocking receptors for aldosterone, not by causing diuresis. This drug and a related agent—eplerenone—are discussed later under “Aldosterone Antagonists.”
Drugs That Inhibit the Renin-Angiotensin-Aldosterone System
The RAAS plays an important role both in cardiac remodeling and in the hemodynamic changes that occur in response to reduced cardiac output. Accordingly, agents that inhibit the RAAS can be highly beneficial. Four groups of drugs are available: ACE inhibitors, ARBs, direct renin inhibitors (DRIs), and aldosterone antagonists. Of the four, the ACE inhibitors have been studied most thoroughly in HF. The basic pharmacology of the RAAS inhibitors is presented in Chapter 36.
Angiotensin-Converting Enzyme Inhibitors
ACE inhibitors (e.g., captopril, enalapril) are a cornerstone of HF therapy. These drugs can improve functional status and prolong life. In one trial, the 2-year mortality rate for patients taking enalapril was 47% lower than the rate for patients taking placebo. Other large, controlled trials have shown similar benefits. Accordingly, in the absence of specific contraindications, all patients with HF should receive one of these drugs. Although ACE inhibitors can be used alone, they are usually combined with a beta blocker and a diuretic.
ACE inhibitors block production of angiotensin II, decrease release of aldosterone, and suppress degradation of kinins. As a result, they improve hemodynamics and favorably alter cardiac remodeling.
Hemodynamic Benefits
By suppressing production of angiotensin II, ACE inhibitors cause dilation of arterioles and veins, and they decrease release of aldosterone. Resulting benefits in HF are as follows:
• Arteriolar dilation improves regional blood flow in the kidneys and other tissues, and, by reducing afterload, it increases stroke volume and cardiac output. Increased renal blood flow promotes excretion of sodium and water.
• Venous dilation reduces venous pressure and thereby reduces pulmonary congestion, peripheral edema, preload, and cardiac dilation.
• Suppression of aldosterone release enhances excretion of sodium and water while causing retention of potassium.
Interestingly, suppression of angiotensin II production diminishes over time, suggesting that long-term benefits are the result of some other action.
Influence on Cardiac Remodeling
With continued use, ACE inhibitors have a favorable influence on cardiac remodeling. Elevation of kinins is largely responsible. This statement is based in part on the observation that, in experimental models, giving a kinin receptor blocker decreases beneficial effects on remodeling. Also, we know that suppression of angiotensin II production diminishes over time, so reduced angiotensin II cannot fully explain long-term benefits.
Adverse Effects
The principal adverse effects of the ACE inhibitors are hypotension (secondary to arteriolar dilation), hyperkalemia (secondary to decreased aldosterone release), intractable cough, and angioedema. In addition, these drugs can cause renal failure in patients with bilateral renal artery stenosis. Because of their ability to elevate potassium levels, ACE inhibitors should be used with caution in patients taking potassium supplements or a potassium-sparing diuretic (e.g., spironolactone, triamterene).
 Black Box Warning: Risk With ACE Inhibitors During Pregnancy
Black Box Warning: Risk With ACE Inhibitors During Pregnancy
Use of ACE inhibitors during pregnancy—especially during the second and third trimesters— can cause fetal injury. Accordingly, if pregnancy occurs, these drugs should be discontinued.
Dosage
Adequate dosage is still debated in the literature: higher dosages may be associated with increased survival, but conflicting evidence remains. Results of the Assessment of Treatment with Lisinopril and Survival (ATLAS) trial indicate that the doses needed to increase survival are higher than those needed to produce hemodynamic changes. In contrast to the ATLAS trial, the NETWORK trial and the High Dose Enalapril Study Group did not find a difference in mortality between patients on low-dose or high-dose ACE inhibitors. Treatment with ACE inhibitors should be initiated at low doses and slowly titrated upward if the patient tolerates treatment. Target dosages associated with disease modification are shown in Table 40.1. These dosages should be used unless side effects make them intolerable.
TABLE 40.1
Inhibitors of the Renin-Angiotensin-Aldosterone System Used in Heart Failure
| Drug | Initial Daily Dose | Maximum Daily Dose |
| ACE INHIBITORS | ||
| Captopril [Capoten] | 6.25 mg 3 times | 50 mg 3 times |
| Enalapril [Vasotec] | 2.5 mg twice | 10–20 mg twice |
| Fosinopril [Monopril] | 5–10 mg once | 40 mg once |
| Lisinopril [Zestril, Prinivil] | 2.5–5 mg once | 20–40 mg once |
| Quinapril [Accupril] | 5 mg twice | 20 mg twice |
| Ramipril [Altace] | 1.25–2.5 mg once | 10 mg once |
| Trandolapril [Mavik] | 1 mg once | 4 mg once |
| ANGIOTENSIN II RECEPTOR BLOCKERS | ||
| Candesartan [Atacand] | 4–8 mg once | 32 mg once |
| Losartan [Cozaar] | 25–50 mg once | 50–100 mg once |
| Valsartan [Diovan] | 20–40 mg twice | 160 mg twice |
| ANGIOTENSIN RECEPTOR NEPRILYSIN INHIBITOR | ||
| Sacubitril/Valsartan [Entresto] | 24/26 mg twice | 97/103 mg twice |
| ALDOSTERONE ANTAGONISTS | ||
| Eplerenone [Inspra] | 25 mg once | 50 mg once |
| Spironolactone [Aldactone] | 12.5–25 mg once | 25 mg once or twice |
Angiotensin II Receptor Blockers
In patients with HF, the effects of ARBs are similar to those of ACE inhibitors—but not identical. Hemodynamic effects of both groups are much the same. Clinical trials have shown that ARBs improve LV ejection fraction, reduce HF symptoms, increase exercise tolerance, decrease hospitalization, enhance quality of life, and, most important, reduce mortality. However, because ARBs do not increase levels of kinins, their effects on cardiac remodeling are less favorable than those of ACE inhibitors. For this reason, and because clinical experience with ACE inhibitors is much greater than with ARBs, ACE inhibitors are generally preferred. For now, ARBs should be reserved for HF patients who cannot tolerate ACE inhibitors, usually owing to intractable cough. (Because ARBs do not increase bradykinin levels, they do not cause cough.)
Angiotensin Receptor Neprilysin Inhibitor
Sacubitril/Valsartan [Entresto]
Sacubitril/valsartan [Entresto] is a newly approved drug that functions in two different manners. Sacubitril is a new class of drug, called an angiotensin receptor neprilysin inhibitor (ANRI). In simple terms, Entresto increases natriuretic peptides while suppressing the negative effects of the RAAS. As discussed earlier in the chapter, ANP and BNP are important indices of cardiac status in HF patients. Entresto is approved for patients with stages II to IV HF to be used in place of an ACE inhibitor or ARB.
In the PARADIGM-HF study, Entresto was superior to enalapril alone when looking at the overall end points of reduction in hospitalizations, risk for all-cause mortality, and risk for death from cardiovascular causes. In fact, the study was terminated early because of the overwhelmingly positive results.
Because Entresto contains an ARB, the contraindications and side effects are similar. Entresto can cause angioedema, hyperkalemia, and hypotension. Administration should be avoided in pregnancy, as use can cause fetal harm.
Aldosterone Antagonists
In patients with HF, aldosterone antagonists—spironolactone [Aldactone] and eplerenone [Inspra]—can reduce symptoms, decrease hospitalizations, and prolong life. These benefits were first demonstrated with spironolactone in the Randomized Aldactone Evaluation Study (RALES). Similar results were later obtained with eplerenone. Current guidelines recommend adding an aldosterone antagonist to standard HF therapy, but only in patients with persistent symptoms despite adequate treatment with an ACE inhibitor and a beta blocker.
Aldosterone antagonists work primarily by blocking aldosterone receptors in the heart and blood vessels. To understand these effects, we need to review the role of aldosterone in HF. In the past, researchers believed that all aldosterone did was promote renal retention of sodium (and water) in exchange for excretion of potassium. However, we now know that aldosterone has additional—and more harmful—effects, including the following:
• Promotion of myocardial remodeling (which impairs pumping)
• Promotion of myocardial fibrosis (which increases the risk for dysrhythmias)
• Activation of the SNS and suppression of norepinephrine uptake in the heart (both of which can promote dysrhythmias and ischemia)
• Promotion of vascular fibrosis (which decreases arterial compliance)
During HF, activation of the RAAS causes levels of aldosterone to rise. In some patients, levels reach 20 times normal. As aldosterone levels grow higher, harmful effects increase, and prognosis becomes progressively worse.
Drugs can reduce the effects of aldosterone by either decreasing aldosterone production or blocking aldosterone receptors. ACE inhibitors, ARBs, and DRIs decrease aldosterone production; spironolactone and eplerenone block aldosterone receptors. Although ACE inhibitors and ARBs can reduce aldosterone production, they do not block it entirely. Furthermore, production is suppressed only for a relatively short time. Hence, when ACE inhibitors or ARBs are used alone, detrimental effects of aldosterone can persist. However, when an aldosterone antagonist is added to the regimen, any residual effects are eliminated. As a result, symptoms of HF are improved and life is prolonged.
Aldosterone antagonists have one major adverse effect: hyperkalemia. The underlying cause is renal retention of potassium. Risk is increased by renal impairment and by using an ACE inhibitor or ARB. To minimize risk, potassium levels and renal function should be measured at baseline and periodically thereafter. Potassium supplements should be discontinued.
Spironolactone—but not eplerenone—poses a significant risk for gynecomastia (breast enlargement) in men, a condition that can be both cosmetically troublesome and painful. In the RALES trial, 10% of males experienced painful breast enlargement.
 Black Box Warning: Spironolactone
Black Box Warning: Spironolactone
Spironolactone is associated with tumorigenesis in studies completed on rats.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


