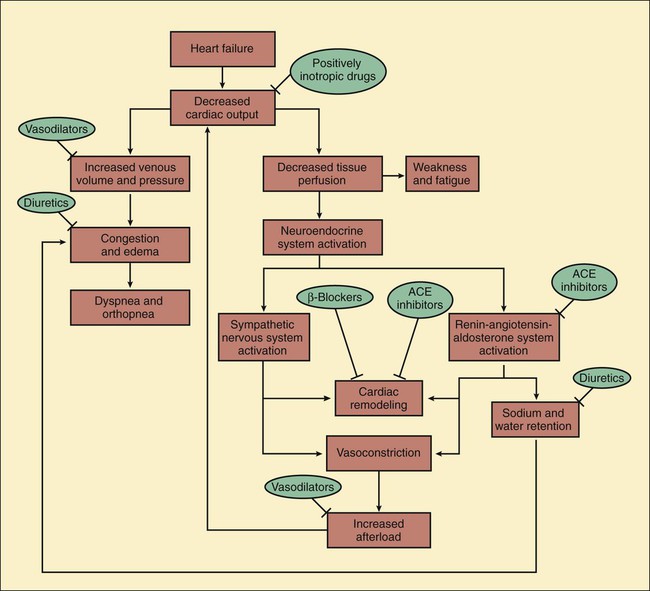
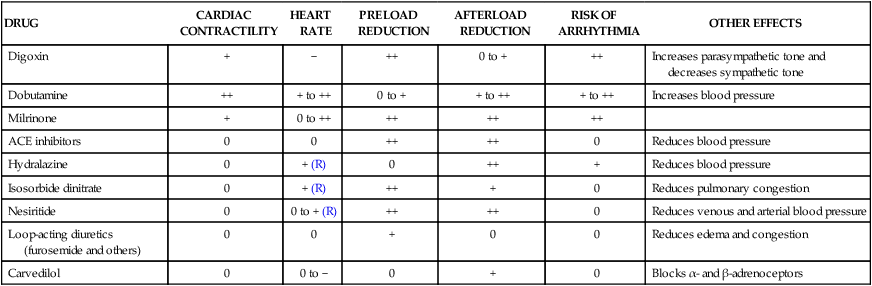
Heart failure is the end stage of a number of cardiovascular disorders that ultimately impair the ability of the ventricle to fill with blood or to eject blood into the circulation. Ischemic heart disease is the most common cause of heart failure. Other important causes of heart failure include hypertension, valvular disorders, arrhythmias, viral and congenital cardiomyopathy, and constrictive pericarditis. Less commonly, heart failure results from severe anemia, thiamine deficiency, or the use of certain anticancer drugs, such as doxorubicin (see Chapter 45). Over time, these disorders produce molecular and cellular changes in cardiac myocytes and connective tissue that lead to a series of structural and functional alterations in the ventricular wall. This process, known as cardiac or ventricular remodeling, is characterized by cardiac dilatation, ventricular wall thinning, interstitial fibrosis, and wall stiffness. These changes impair the ability of the heart to relax or contract. In cases of left ventricular failure (left-sided heart failure), the left ventricle does not adequately pump blood forward, so the pressure in the pulmonary circulation increases. When the increased pressure forces fluid into the lung interstitium, this causes congestion and edema (Fig. 12-1). Pulmonary edema reduces the diffusion of oxygen and carbon dioxide between alveoli and the pulmonary capillaries. This causes hypoxemia (deficient oxygenation of the blood) and can lead to dyspnea (difficulty in breathing), including exertional dyspnea (dyspnea provoked by exercise), orthopnea (intensified dyspnea when lying flat), and paroxysmal nocturnal dyspnea (edema-induced bronchoconstriction when sleeping). The reduction in cardiac output that occurs in heart failure triggers a cascade of compensatory neuroendocrine responses. Although these responses attempt to restore cardiac output via the Frank-Starling mechanism (see Fig. 12-1), they are often maladaptive and counterproductive. The reduction in tissue perfusion activates both the sympathetic nervous system and the renin-angiotensin-aldosterone system, both of which in turn stimulate vasoconstriction. Arterial vasoconstriction increases aortic impedance to left ventricular ejection and thereby decreases cardiac output, especially in patients with a weak, dilated heart. When angiotensin II stimulates the secretion of aldosterone and antidiuretic hormone, this increases the amount of sodium and water retention, the plasma volume, and the venous pressure. In addition, angiotensin II and sympathetic activation lead to cardiac remodeling and ventricular wall thinning or fibrosis, which often reduce systolic and diastolic function. Hence, the net result of the neuroendocrine responses is often a further reduction in cardiac output and an increase in circulatory congestion. Table 12-1 compares the cardiovascular effects of drugs discussed in this chapter. Each of these drugs partly counteracts the loss of myocardial function and the maladaptive responses that occur during heart failure; however, none of the current therapies, either alone or in combination, has been completely satisfactory. Because heart failure has such a high incidence and poor prognosis, a much greater effort has been expended in the search for better means to treat it. The most significant development in recent decades has been the use of angiotensin inhibitors, β-adrenoceptor blockers, and other agents that attenuate cardiac remodeling and reduce the mortality rate in patients with heart failure. Ultimately, however, the successful treatment of patients with heart failure may require the development of drugs that activate genes capable of repairing or replacing myocardial tissue. TABLE 12-1 Cardiovascular Effects of Drugs Used in the Treatment of Heart Failure* ACE, Angiotensin converting enzyme. *Effects are indicated as follows: decrease (−); no change or variable (0); increase ranging from small (+) to large (++); and reflex (R). As shown in Table 12-2, digoxin is adequately absorbed from the gut and has a long half-life of about 36 hours. It is primarily eliminated by renal excretion of the parent compound. Because digoxin has a low therapeutic index, serum concentrations are useful in assessing the adequacy of the dosage and evaluating potential toxicity and should be in the range of 0.5 to 2 ng/mL. TABLE 12-2 Pharmacokinetic Properties of Positively Inotropic Drugs*
Drugs for Heart Failure
Overview
Pathophysiology of Heart Failure
Mechanisms and Effects of Drugs for Heart Failure
DRUG
CARDIAC CONTRACTILITY
HEART RATE
PRELOAD REDUCTION
AFTERLOAD REDUCTION
RISK OF ARRHYTHMIA
OTHER EFFECTS
Digoxin
+
−
++
0 to +
++
Increases parasympathetic tone and decreases sympathetic tone
Dobutamine
++
+ to ++
0 to +
+ to ++
+ to ++
Increases blood pressure
Milrinone
+
0 to ++
++
++
++
ACE inhibitors
0
0
++
++
0
Reduces blood pressure
Hydralazine
0
+ (R)
0
++
+
Reduces blood pressure
Isosorbide dinitrate
0
+ (R)
++
+
0
Reduces pulmonary congestion
Nesiritide
0
0 to + (R)
++
++
0
Reduces venous and arterial blood pressure
Loop-acting diuretics (furosemide and others)
0
0
+
0
0
Reduces edema and congestion
Carvedilol
0
0 to −
0
+
0
Blocks α- and β-adrenoceptors
Positively Inotropic Drugs
Digoxin
Drug Properties
Chemistry and Pharmacokinetics
DRUG
ORAL BIOAVAILABILITY
ONSET OF ACTION
DURATION OF ACTION
ELIMINATION HALF-LIFE
EXCRETED UNCHANGED IN URINE
THERAPEUTIC SERUM LEVEL
Digoxin
75%
1 hr
24 hr
36 hr
60%
0.5-2 ng/mL
Dobutamine
NA
1 min
<10 min
2 min
0%
NA
Milrinone
NA
3 min
Variable
4 hr
60%
NA ![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree