Chapter 11 Drugs Affecting the Sympathetic Nervous System
| Therapeutic Overview |
|---|
| Mpathomimetics |
| Nasal decongestion |
| Decrease formation of aqueous humor in glaucoma |
| Neurogenic shock, cardiogenic shock |
| Paroxysmal atrial tachycardia |
| Bronchospasm, asthma |
| Decrease diffusion of local anesthetics |
| Sympatholytics |
| Essential hypertension, hypertensive emergencies |
| Benign prostatic hyperplasia |
| Cardiac dysrhythmias, angina pectoris, postmyocardial infarction |
| Essential tremor |
| Glaucoma |
Therapeutic Overview
The Therapeutic Overview Box presents a summary of the primary uses of different classes of compounds that affect the sympathetic nervous system.
Mechanisms of Action
The biochemistry and physiology of the autonomic nervous system, including a discussion of adrenergic receptors, is presented in Chapter 9. Detailed information on receptors and signaling pathways involved are in Chapter 1. This section covers topics that pertain specifically to noradrenergic neurotransmission and its modulation by drugs.
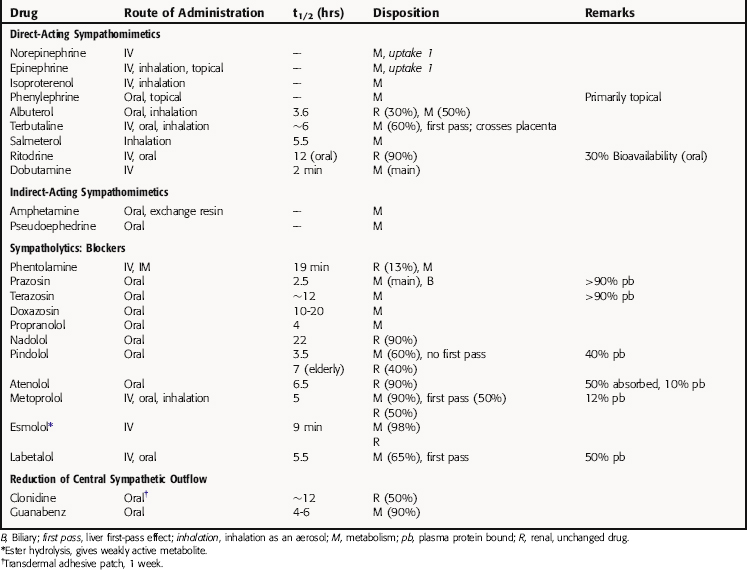
The neurochemical steps that mediate noradrenergic neurotransmission are summarized in Figure 11-1. The first step involves the transport of tyrosine into the neuron followed by its conversion to 4-dihydroxy-phenylalanine (l-DOPA) by the rate-limiting enzyme tyrosine hydroxylase. l-DOPA is rapidly decarboxylated to dopamine (DA) by aromatic l-amino acid decarboxylase (ALAAD), also known as DOPA decarboxylase. In dopaminergic neurons within the central nervous system (CNS), this is the last step in the synthetic process, and DA is released as a neurotransmitter. Noradrenergic neurons in the CNS and peripheral sympathetic nervous system contain an additional enzyme, DA β-hydroxylase, which converts DA to NE. This enzyme is located in synaptic vesicles, so DA must be actively transported into these vesicles before conversion to NE.
When the action potential depolarizes the nerve terminal, voltage-gated Ca++ channels open, allowing Ca++ to enter the neuron and cause the vesicles to release NE. At the neuroeffector junction, NE binds to adrenergic receptor subtypes on both the presynaptic and postsynaptic membranes, with different organs containing different receptor subtypes. Activation of α2-“autoreceptors” on the presynaptic neuronal membrane causes feedback inhibition and reduces further NE release. The signaling mechanisms occurring after adrenergic receptor activation are discussed in Chapter 9.
After NE has activated its receptors, its action is terminated primarily by a high-affinity reuptake system (termed “uptake 1” in Fig. 11-1), which transports NE back into the neuron for repackaging and eventual re-release. A smaller fraction of the NE in the synapse diffuses away from the receptors and can be taken up by a lower affinity extraneuronal process (termed “uptake 2” in Fig. 11-1). Some of the NE taken back into the sympathetic neurons by reuptake can be oxidatively deaminated by monoamine oxidase (MAO) located on the external mitochondrial membrane. The biologically inactive deaminated metabolites enter the circulation and are excreted in the urine. The NE that is transported into the postjunctional cell is O-methylated by catechol-O-methyltransferase (COMT) to normetanephrine. The high-affinity reuptake of NE into presynaptic terminals is the major mechanism that terminates its actions, although COMT and MAO also play important roles in metabolizing circulating NE and Epi and some exogenously administered sympathomimetic amines.
Drugs modify the activity of sympathetic neurons by increasing or decreasing the noradrenergic signal (see Fig. 11-1). Sympathomimetics may mimic noradrenergic transmission by acting directly on postsynaptic adrenergic receptors (e.g., phenylephrine), by facilitating NE release (e.g., amphetamine) or by blocking neuronal reuptake (e.g., cocaine). Amphetamine and cocaine have intense effects on the CNS and are drugs of abuse with severe addiction liability (Chapter 37).
Activation of Adrenergic Receptors
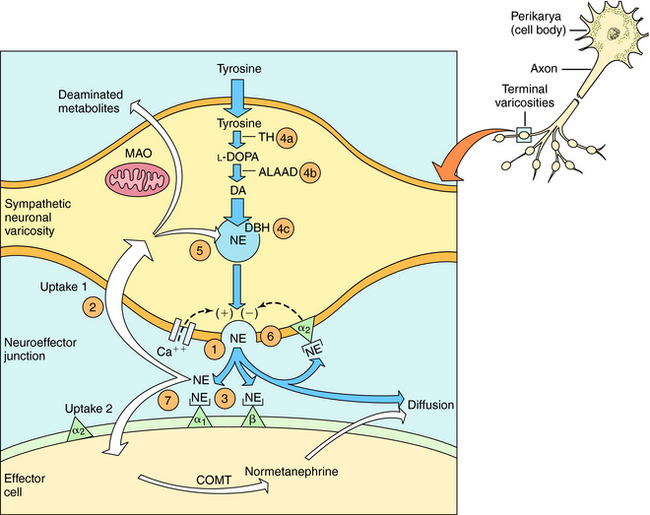
Over the last half century it has become clear that the actions of NE and Epi are mediated through multiple adrenergic receptor subtypes (see Chapter 9). We now know that there are nine different subtypes of adrenergic receptors, each encoded by separate genes, which are grouped into three major families (α1, α2, β), each containing three different members. Of these, only four (α1, α2, β1, and β2) are currently important in clinical pharmacology; therefore they are the major focus of this chapter. Both NE and Epi activate most adrenergic receptors with similar, but not identical, potencies, although NE is much less potent than Epi at the β2-subtype. Isoproterenol (ISO) is a synthetic analog of NE and Epi and selectively activates only β receptors. Differences in the pharmacological profiles of different adrenergic receptors are illustrated in Figure 11-2, where dose-response curves for the actions of these catecholamines on different tissues are depicted.
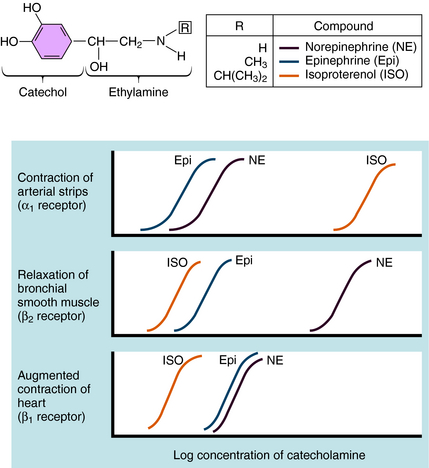
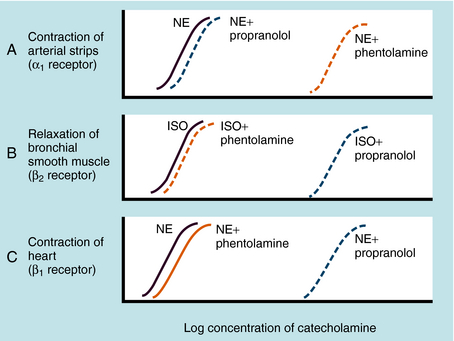
Adrenergic receptor activation increases the contraction of cardiac muscle and induces smooth muscle to either contract or relax, depending on the receptor subtype(s) present. Contraction of arterial strips is mediated by α1 receptors, where the relative potencies of the three catecholamines are Epi ≥ NE >>> ISO. Relaxation of bronchial smooth muscle is mediated by β2 receptors, with the relative potencies being ISO > Epi >>> NE. Contraction of cardiac muscle is mediated by β1 receptors, with the relative potencies being ISO > Epi = NE. The presence of different receptor subtypes in these three tissues is further demonstrated in Figure 11-3 by examining the effects of selective antagonists. Phentolamine, a competitive antagonist at α1 receptors, causes a parallel shift to the right of NE-induced contractions of arterial strips (see Fig. 11-3, A) but does not affect the other two responses. Propranolol, a competitive antagonist at both β1 and β2 receptors, causes a parallel shift to the right of responses mediated by bronchial β2 receptors (see Fig. 11-3, B) and cardiac β1 receptors (see Fig. 11-3, C), without affecting the α1 receptor response.
Adrenergic α2 receptors are found on platelets, in the CNS, and postsynaptically in several other peripheral tissues (blood vessels, pancreas, and enteric cholinergic neurons). As mentioned, activation of α2 receptors on the terminals of sympathetic neurons reduces NE release. These receptors are also found both presynaptically and postsynaptically on neurons in brain, and activation of these receptors reduces central sympathetic outflow.
Direct-Acting Sympathomimetics
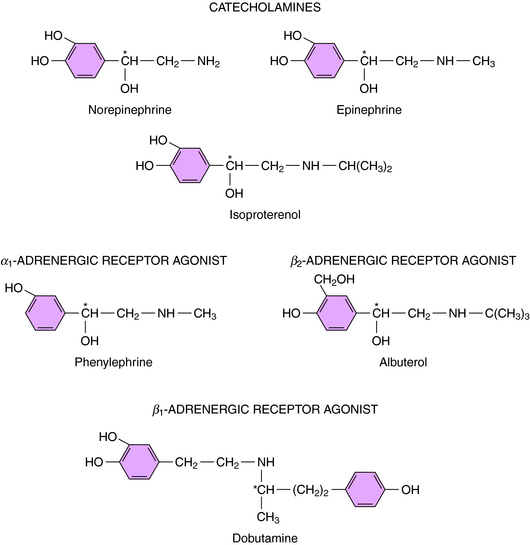
Direct-acting adrenergic receptor agonists mimic some of the effects of sympathetic nervous system activation by binding to and activating specific receptor subtypes (see Fig. 11-1, site 3). For example, as mentioned, ISO selectively activates β receptors, phenylephrine selectively activates α1 receptors, and clonidine selectively activates α2 receptors. Similarly, dobutamine and albuterol are relatively specific agonists at β1 and β2 receptors, respectively. The structures of some clinically important adrenergic receptor agonists are shown in Figure 11-4.
Indirect-Acting Sympathomimetics
Indirect-acting sympathomimetics do not activate receptors directly but facilitate the release of NE from sympathetic neurons or block high-affinity reuptake. Thus they act presynaptically to facilitate adrenergic neurotransmission. Amphetamine and related drugs produce their sympathomimetic effects by facilitating NE release (see Fig. 11-1, site 1); the effects of these drugs in the CNS are described in Chapter 37. On the other hand, cocaine and tricyclic antidepressants, such as desipramine, exert their sympathomimetic effects by blocking high-affinity reuptake (see Fig. 11-1, site 2). The structure and characteristics of cocaine are discussed in Chapter 37, with similar information for tricyclic antidepressants given in Chapter 30.
Because neuronal reuptake, and not metabolism, is the primary mechanism for terminating the actions of NE and Epi, it is not surprising that drugs that inhibit the metabolism of these amines show little or no sympathomimetic actions. On the other hand, inhibitors of MAO (e.g., pargyline) or COMT (e.g., tolcapone) can enhance the actions of other synthetic sympathomimetic amines that are substrates for these enzymes, with some important toxicological implications. For example, the actions of tyramine, a sympathomimetic amine present in a variety of foods, are greatly enhanced in patients treated with MAO inhibitors (see Chapter 30), and the pharmacokinetics of l-DOPA, used for the treatment of Parkinson’s disease, are increased when administered concurrently with a COMT inhibitor (see Chapter 28).
An indirect-acting sympathomimetic amine that releases NE must penetrate the noradrenergic neuron before it can act. Nonpolar, lipid-soluble drugs such as amphetamine can diffuse across neuronal membranes, whereas polar, water-soluble compounds such as tyramine must rely on high-affinity uptake by the NE transporter. Thus drugs that inhibit the NE transporter will reduce or block the effects of tyramine but will enhance the effects of Epi, which acts directly on the adrenergic receptors. Cocaine also causes a prompt increase in the response to Epi by blocking the reuptake system, whereas reserpine disrupts amine transport from the cytoplasm into synaptic vesicles (see Fig. 11-1, site 5). Therefore reserpine has only small effects on responses to direct-acting sympathomimetics.
Inhibition of Synthesis, Storage, or Release of NE
Catecholamine synthesis can be disrupted at several steps, but effective in vivo blockade is obtained only when tyrosine hydroxylase, the enzyme catalyzing the first and rate-limiting step, is inhibited (see Fig. 11-1, site 4a). This can be produced clinically with α-methyltyrosine (metyrosine). Several compounds can inhibit other biosynthetic enzymes (e.g., DOPA decarboxylase is inhibited by carbidopa and DA β-hydroxylase is inhibited by disulfiram (see Fig. 11-1, sites 4b and 4c, respectively). Although these drugs do not effectively block endogenous catecholamine synthesis when administered, they do have clinical utility by affecting other systems. By virtue of its ability to inhibit peripheral DOPA decarboxylase, carbidopa is used in combination with l-DOPA for patients with Parkinson’s disease, to increase the amount of l-DOPA available to the CNS to enhance DA synthesis (see Chapter 28). Disulfiram is used in treating chronic alcoholism (see Chapter 32) because it blocks aldehyde dehydrogenase; however, its ability to inhibit catecholamine synthesis leads to significant adverse effects such as hypotension.
Disruption of vesicular storage also modifies noradrenergic transmission (see Fig. 11-1, site 5). As discussed, reserpine disrupts the ability of the synaptic vesicles to transport and store DA and NE. Adrenergic responses can also be impeded by inhibiting NE release (see Fig. 11-1, site 6). Drugs such as bretylium and guanethidine, which accumulate in noradrenergic nerve terminals, prevent NE release.
Adrenergic Receptor Antagonists
Many adrenergic receptor antagonists exert different subtype-selective effects (see Fig. 11-1, site 7). The early antagonists such as phenoxybenzamine and propranolol blocked α or β receptors, respectively, whereas drugs are now available that selectively block α1 (prazosin), α2 (yohimbine), or β1 (metoprolol) receptors. These selective antagonists have important therapeutic advantages over the original broad-spectrum adrenergic receptor blockers and are used for various indications, such as the control of blood pressure (see Chapter 20).