Chapter 10 Drugs Affecting the Parasympathetic Nervous System and Autonomic Ganglia
| Abbreviations | |
|---|---|
| AcCoA | Acetyl coenzyme A |
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| ATP | Adenosine triphosphate |
| BuChE | Butyrylcholinesterase |
| ChAT | Choline acetyltransferase |
| ChE | Cholinesterase |
| CNS | Central nervous system |
| COPD | Chronic obstructive pulmonary disease |
| Epi | Epinephrine |
| EPSP | Excitatory postsynaptic potential |
| GI | Gastrointestinal |
| NO | Nitric oxide |
| 2-PAM | Pralidoxime |
| PNS | Peripheral nervous system |
| SNP | Single-nucleotide polymorphism |
| VEGF | Vascular endothelial growth factor |
Therapeutic Overview
Drugs that block the actions of ACh at muscarinic receptors are termed muscarinic receptor antagonists, parasympatholytics, or cholinolytics, and are used to treat motion sickness, relieve some of the symptoms of Parkinson’s disease (see Chapter 28), dilate the pupils for ocular examination, reduce motility of the GI and urinary tracts, dilate the airways of the lung in patients with chronic obstructive pulmonary disease (COPD)
| Therapeutic Overview |
|---|
| Muscarinic Receptor Agonists |
| Glaucoma |
| Postoperative ileus, congenital megacolon, and urinary retention |
| Cholinesterase (ChE) Inhibitors |
| Glaucoma |
| Postoperative ileus, congenital megacolon, urinary retention |
| Diagnosis and treatment of myasthenia gravis |
| Reversal of neuromuscular blockade following surgery |
| Alzheimer’s disease |
| Muscarinic Receptor Antagonists |
| Motion sickness |
| Examination of the retina and measurement of refraction; inflammatory uveitis |
| Excessive motility of GI and urinary tract; urinary incontinence; irritable bowel syndrome |
| Chronic obstructive pulmonary disease |
| Parkinson’s disease |
| Ganglionic Blockers |
| Hypertensive emergencies |
(see Chapter 16), and reduce acid secretion in individuals with peptic ulcer disease (see Chapter 18).
Drugs that antagonize the actions of ACh at ganglionic nicotinic receptors are referred to as ganglionic blockers and are used to treat hypertensive emergencies. Drugs that antagonize the actions of ACh at nicotinic receptors at the neuromuscular junction are termed neuromuscular blocking agents and are used to relax skeletal muscle (see Chapter 12).
Mechanisms of Action
The biochemistry and physiology of the autonomic nervous system, including a discussion of muscarinic and nicotinic cholinergic receptors, are presented in Chapter 9. Detailed information on receptors and signaling pathways are discussed in Chapter 1. This section covers topics that pertain specifically to cholinergic neurotransmission and its modulation by drugs.
The neurochemical steps that mediate cholinergic neurotransmission are summarized in Figure 10-1. ACh is synthesized from choline and acetyl coenzyme A (AcCoA) by the enzyme choline acetyltransferase (ChAT). ChAT is not rate-limiting for ACh synthesis, and thus ChAT inhibitors have little or no effect on ACh concentrations. The AcCoA used for ACh synthesis is synthesized in mitochondria from its immediate precursor, pyruvate, and is transported out of mitochondria into the cytoplasm.

Once formed in the cytosol, ACh is transported actively into synaptic vesicles by the vesicular ACh transporter. The arrival of an action potential at the nerve terminal causes Ca++ influx, triggering the release of ACh from storage vesicles. The toxin from Clostridium botulinum (botulinum toxin) prevents the exocytotic release of ACh from synaptic vesicles and blocks cholinergic neurotransmission. This mechanism underlies the clinical and cosmetic use of botulinum toxin in conditions that involve involuntary skeletal muscle activity or increased muscle tone including strabismus, blepharospasm, hemifacial spasm, and facial wrinkles (see Chapter 12). After release from the nerve terminal, ACh elicits cellular responses by activating postsynaptic muscarinic or nicotinic receptors. Muscarinic receptors are also located presynaptically, where they inhibit further neurotransmitter release. The effects of ACh are rapidly terminated by the enzyme acetylcholinesterase (AChE), which hydrolyzes ACh to acetate and choline. Choline is transported back into nerve terminals by the high-affinity choline transporter, where it can be reused for ACh synthesis.
Drugs can modify the effectiveness of parasympathetic nerve activity by increasing or decreasing cholinergic neurotransmission. Parasympathomimetics mimic cholinergic transmission by acting directly on postsynaptic receptors (muscarinic receptor agonists) or by prolonging the action of released ACh (AChE inhibitors). Drugs that block cholinergic transmission inhibit either the storage of ACh (vesamicol), vesicular exocytosis (botulinum toxin), or the high-affinity transport of choline (hemicholinium). Drugs can also block muscarinic receptors (muscarinic receptor antagonists). The sites of action of all these compounds are depicted in Figure 10-1.
Activation of Cholinergic Receptors
Drugs that activate cholinergic receptors may be classified as either directly or indirect-acting. Direct-acting compounds produce their effects by binding to and activating either muscarinic or nicotinic receptors and are termed muscarinic or nicotinic receptor agonists, respectively. Indirect-acting cholinomimetics produce their effects by inhibiting AChE, thereby increasing the concentration and prolonging the action of ACh at cholinergic synapses. The directly and indirect-acting cholinomimetics and their clinical uses are presented in Table 10-1.
TABLE 10–1 Direct- and Indirect-Acting Cholinomimetic Drugs and their Uses
| Action | Agent | Use |
|---|---|---|
| Direct-Acting (Muscarinic Receptor Agonists) | ||
| Pilocarpine | Glaucoma | |
| Carbachol | Glaucoma | |
| Bethanechol | Postoperative ileus, congenital megacolon, urinary retention | |
| ChE Inhibitors* | ||
| Physostigmine | Glaucoma | |
| Demecarium | Glaucoma | |
| Echothiophate | Glaucoma | |
| Isoflurophate | Glaucoma | |
| Neostigmine | Postoperative ileus, congenital megacolon, urinary retention, reversal of neuromuscular blockade | |
| Edrophonium | Diagnosis of myasthenia gravis, reversal of neuromuscular blockade, supraventricular tachyarrhythmias | |
| Ambenonium | Treatment of myasthenia gravis | |
| Pyridostigmine | Treatment of myasthenia gravis, reversal of neuromuscular blockade | |
* ChE inhibitors used for Alzheimer’s disease are listed in Chapter 28.
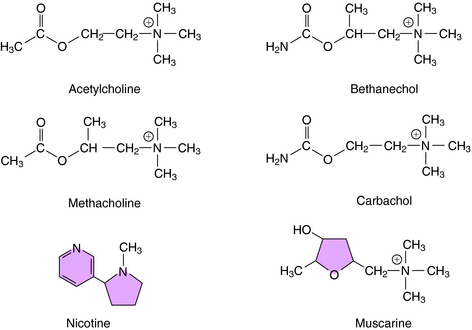
The structures of several direct-acting cholinergic agonists are shown in Figure 10-2. Introduction of a methyl group in the β position of ACh yields agonists selective for muscarinic receptors such as methacholine and bethanechol. In addition, substitution of an amino group for the terminal methyl group yields corresponding carbamic acid ester derivatives (i.e., carbachol and bethanechol), which render these compounds relatively resistant to hydrolysis by AChE, prolonging their action as compared with ACh.
Muscarinic receptors mediate cellular responses by interacting with heterotrimeric G proteins to affect ionic conductances and the cytosolic concentration of second messengers (see Chapters 1 9). As indicated in Chapter 9, five muscarinic receptor subtypes (M1 to M5) have been identified, with M1 receptors located in the CNS, peripheral neurons, and gastric parietal cells, M2 receptors located in the heart and on some presynaptic nerve terminals in the periphery and CNS, and M3 receptors located on glands and smooth muscle. M4 and M5 receptors are located in the CNS and are less well understood than the others.
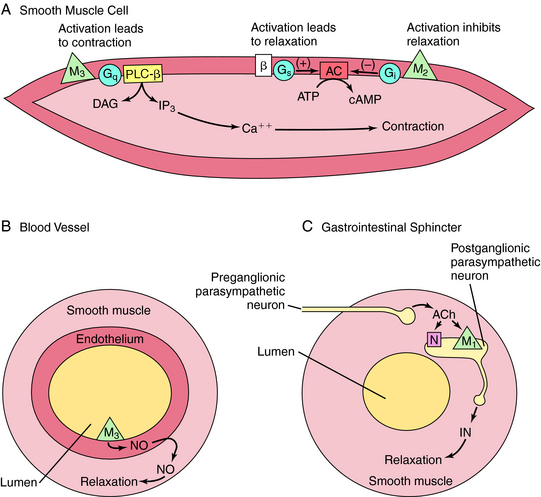
M1, M3, and M5 receptors are coupled to Gq proteins, and stimulation of these receptors activates phospholipase C, leading to increased phosphoinositide hydrolysis and the release of intracellular Ca++. In contrast, M2 and M4 receptors are coupled to Gi, and agonist activation leads to inhibition of adenylyl cyclase. The signaling pathways mediating the contraction of smooth muscle upon activation of M2 or M3 receptors is depicted in Figure 10-3, A. Activation of M3 receptors promotes smooth muscle contraction directly, whereas activation of M2 receptors promotes contraction by inhibiting the ability of β adrenergic receptor activation to relax smooth muscle. Activation of M3 receptors on the endothelium of blood vessels (see Fig. 10-3, B) increases the production of nitric oxide (NO), which diffuses and causes relaxation of the muscle, while activation of M1 receptors in parasympathetic ganglia (see Fig. 10-3, C) releases an inhibitory neurotransmitter such as adenosine triphosphate (ATP), NO, or vasoactive intestinal peptide to relax smooth muscle sphincters.
Most, but not all, peripheral effects of muscarinic receptor agonists resemble those elicited by the activation of parasympathetic nerves (see Chapter 9). These effects include: (1) constriction of the iris sphincter; (2) contraction of the ciliary muscle, resulting in accommodation of the lens; (3) constriction of the airways; (4) an increase in GI motility; (5) contraction of the bladder reservoir and relaxation of its outlet; (6) a decrease in heart rate; and (7) an increase in secretions of sweat glands, salivary glands, lacrimal glands, and glands of the trachea and GI mucosa. Although the ventricular myocardium and most peripheral blood vessels lack cholinergic innervation, they express muscarinic receptors. When activated by exogenously administered agonists, these receptors decrease the force of contraction in the heart and dilate peripheral blood vessels. Muscarinic receptor agonists also activate receptors throughout the CNS, and muscarinic receptors in the brain play an important role in learning, memory, control of posture, and temperature regulation. Excessive activation of central muscarinic receptors causes tremor, convulsions, and hypothermia.
The prototypic nicotinic receptor agonist, nicotine, is found in tobacco, cigarette smoke, some insecticides, and in transdermal patches used to ease withdrawal from smoking. Lobeline is another natural nicotinic receptor agonist that has actions similar to nicotine. The cholinomimetic carbachol has nicotinic as well as muscarinic receptor agonist activity. Nicotine activates nicotinic receptors at the neuromuscular junction, causing contraction followed by depolarization blockade and paralysis. Nicotine also activates sympathetic and parasympathetic ganglia. As a consequence, because of differences in the predominant tone of autonomic systems (see Chapter 9), its effects on the vascular system are primarily sympathetic, whereas its effects on the GI tract are primarily parasympathetic. Nicotine elevates blood pressure and heart rate, with the latter opposed by reflex bradycardia. The sympathetic effects of nicotine are reinforced by stimulation of nicotinic receptors at the adrenal medulla, leading to the release of catecholamines. Nicotine also stimulates the GI and urinary tracts, causing diarrhea and urination. Nicotine readily enters the brain, and increasing doses cause alertness, vomiting, tremors, convulsions, and, ultimately, coma, and is a source of poisoning, especially in children.
Two types of cholinesterase (ChE) enzymes are expressed in the body, AChE and butyrylcholinesterase (BuChE, also called plasma or pseudoChE), each with a different distribution and substrate specificity. AChE is located at synapses throughout the nervous system and is responsible for terminating the action of ACh. BuChE is located at non-neuronal sites, including plasma and liver, and is responsible for the metabolism of certain drugs, including ester-type local anesthetics (Chapter 13) and succinylcholine (Chapter 12). Most enzyme inhibitors used clinically do not discriminate between the two types of ChEs.
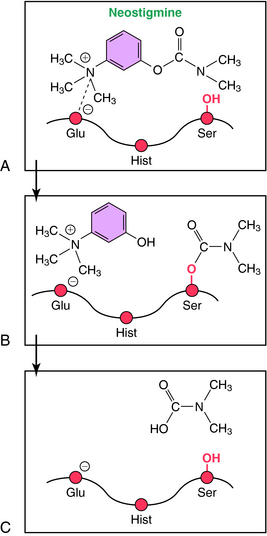
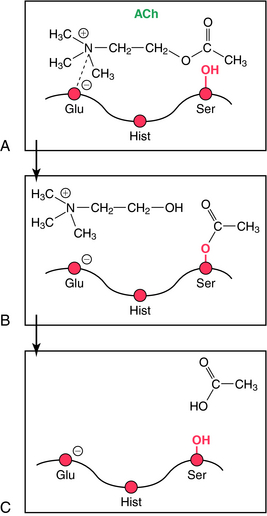
The mechanism involved in the hydrolysis of ACh explains the biochemical basis of the actions of ChE inhibitors (Fig. 10-4). AChE is a member of the serine hydrolase family of enzymes and contains a serine (ser)-glutamate (glu)-histidine (hist) triad at the active site. The enzyme has an anionic and a catalytic domain. When ACh binds to AChE, the quaternary nitrogen of ACh binds to the anionic site, positioning the ester group near the catalytic site. The ester moiety undergoes a nucleophilic attack by the serine of the catalytic site, resulting in the hydrolysis of ACh and binding of acetate to the serine of the catalytic domain (acetylation). The acetylated serine is rapidly hydrolyzed, and the free enzyme is regenerated. This enzymatic reaction is one of the fastest known; approximately 104 molecules of ACh are hydrolyzed per second by a single AChE molecule, requiring only about 100 μsec.
ChE inhibitors are divided into two main types, reversible and irreversible. Reversible inhibitors are further subdivided into noncovalent and covalent enzyme inhibitors. Noncovalent inhibitors, such as edrophonium, bind reversibly to the anionic domain of AChE. The duration of action is determined in part by the way in which the inhibitor binds. Edrophonium binds weakly and has a rapid renal clearance, resulting in a brief duration of action (approximately 10 minutes). Tacrine and donepezil are also noncovalent ChE inhibitors used to treat Alzheimer’s disease (Chapter 28); they have higher affinities and partition into lipids, giving longer durations of action.
Covalent reversible ChE inhibitors, such as physostigmine and neostigmine, are sometimes referred to as carbamate inhibitors and are carbamic acid ester derivatives. These compounds bind to AChE and are hydrolyzed, but at a relatively slow rate (Fig. 10-5). The resultant carbamylated enzyme is more stable than the acetylated enzyme produced by the hydrolysis of ACh. Unlike the acetylated enzyme, which is deacetylated within seconds, the carbamylated enzyme takes 3 to 4 hours to decarbamylate, contributing to the moderate duration of action of these compounds in patients.