INTRODUCTION
Over the past decade, the US Food and Drug Administration (FDA) has approved approximately 260 new therapeutics, including 220 new molecular entities (small molecules) and 42 biotherapeutics (generally, recombinant protein products). Many such therapies have enabled treatments for diseases that were previously untreatable. Others have yielded expanded treatment options because they are more efficacious and/or less toxic than previously available treatments. In the fight against infectious diseases, for example, pharmaceutical and biotechnology companies, university laboratories, and others have continued to develop new agents to treat diseases that have become resistant to existing treatments. With the availability of new technologies such as next-generation sequencing and novel protein engineering strategies, it is anticipated that important new classes of therapeutics will continue to be discovered and developed in the coming decades.
The development of a new drug is difficult and costly. Very few molecules that reach the development stage are ultimately approved as drugs: of 10,000 chemical compounds considered promising from the results of initial screening assays, fewer than 10 make it to clinical trials and only 2 are eventually approved. Furthermore, the costs associated with discovering and developing a new drug are estimated to be slightly over $1.2 billion, with some estimates suggesting costs of up to $5 billion. Although the development of new drugs is a risky venture, successful drugs can be quite profitable for those willing to take such risks. The biggest commercial successes, such as Abilify®, have annual sales of more than $6 billion each.
Increased attention has recently been focused on the inability of the biomedical research community to produce innovative new therapies. The challenges involved in drug discovery and development were highlighted (along with potential solutions) in the 2004 report of the FDA Critical Path Initiatives (see “Suggested Reading”). This report noted that both the National Institutes of Health (NIH) budget and pharmaceutical company research and development spending approximately doubled over a 10-year period beginning in 1993. The added investment did not increase the rate of development of new medicines, however, as evidenced by a decline in major drug and biological product submissions to the FDA. While several potential solutions have been offered to address this issue, it is important to note that a joint report by the FDA and the Association of American Medical Colleges highlighted the critical role of physician-scientists in improving the effectiveness of drug discovery and development.
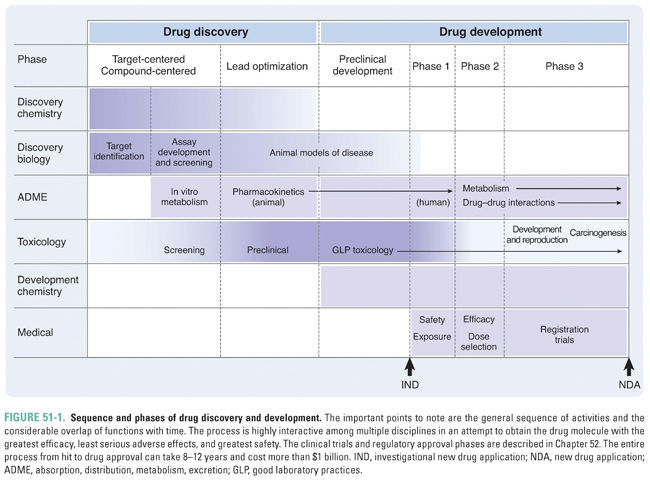
This chapter describes the phases of drug discovery and development and the scientific disciplines that are involved in these phases. Drug discovery spans the period from the identification of a potential therapeutic target to the selection of molecules for testing in humans. Drug development is generally defined as the period from the preclinical studies that support initial clinical trials through approval of the drug by regulatory authorities. The process of drug discovery and development is complex, requiring contributions from many otherwise disparate scientific disciplines (Fig. 51-1).
 Drug discovery programs often start with a well-characterized biochemical or molecular target that is known to mediate a disease of interest. Drug discovery then involves the pursuit of a chemical compound (small molecule) or engineered macromolecule (generally, a protein) designed to modulate the pathway of interest. The following case provides an example of an alternative approach, in which an extensive genomics and bioinformatics program led to the identification of a novel protein (osteoprotegerin) and the discovery of a new pathway important in bone metabolism. These basic science discoveries then led to the development and commercialization of a novel protein therapeutic that is currently used to treat osteoporosis and bone destruction associated with cancer metastasis.
Drug discovery programs often start with a well-characterized biochemical or molecular target that is known to mediate a disease of interest. Drug discovery then involves the pursuit of a chemical compound (small molecule) or engineered macromolecule (generally, a protein) designed to modulate the pathway of interest. The following case provides an example of an alternative approach, in which an extensive genomics and bioinformatics program led to the identification of a novel protein (osteoprotegerin) and the discovery of a new pathway important in bone metabolism. These basic science discoveries then led to the development and commercialization of a novel protein therapeutic that is currently used to treat osteoporosis and bone destruction associated with cancer metastasis.
In the early 1990s, researchers at Amgen engaged in an extensive genomics program designed to identify novel genes and proteins in the anticipation that new biologic pathways and potential therapeutic targets would be identified. Full-length sequences were generated for specific genes of interest and were overexpressed in the livers of mice. These transgenic mice were then used in a phenotypic screen to identify biochemical, hematologic, radiographic, and histologic differences from wild-type control mice. Priority was given to genes encoding secreted proteins and genes encoding new members of families of proteins already known to have important roles in disease pathways. Through this process, investigators identified osteoprotegerin (OPG), a new protein with sequence homology to members of the tumor necrosis factor receptor (TNFR) family. Mice overexpressing OPG had a striking bone phenotype, with marked increases in bone that filled the medullary cavity. This increase in bone density was attributed to a marked reduction in osteoclast number. In 1995, Amgen patent filings identified OPG as an important regulator of bone metabolism. Starting from this discovery of OPG, investigators from Amgen and other laboratories elucidated a novel and critical pathway in osteoclast biology, including the discovery of receptor activator of NF-κB (RANK) and its ligand (RANKL).
Initial strategies to convert this biologic understanding into a useful therapeutic focused on OPG, including a fusion protein of OPG combined with a human immunoglobulin G1 (IgG1) Fc region. In animal models, this fusion protein was 200 times more active than full-length OPG, and, following safety testing in animals, it entered clinical trials in 1998. The initial Phase 1 study demonstrated dose-related reductions in bone turnover markers and provided proof of concept that modulation of the RANKL pathway could have beneficial effects on the human skeleton. Further optimization efforts led to the development of a similar molecule, derived from a mammalian cell line, with increased target affinity and a longer half-life. This molecule (AMGN-0007) entered clinical development; however, its development was halted based on the induction of an immune response to OPG in a clinical trial subject. Because of concerns regarding the potential for an OPG construct to induce an immune response and neutralize endogenous OPG, efforts to develop an OPG-based drug were discontinued.
Amgen had not limited its research program to an OPG-based therapeutic but had also capitalized on the significant progress that had been made in the area of monoclonal antibody generation technology to initiate development of a fully human monoclonal antibody targeting RANKL. These efforts led to the identification of AMG 162 (later known as denosumab). Because denosumab was not active in rodent species, studies in nonhuman primates were essential to demonstrate that denosumab had beneficial effects on the skeleton. Studies in nonhuman primates were also used to characterize the toxicity profile of denosumab, after which the drug entered clinical trials for the indication of osteoporosis in 2001. In the Phase 1 clinical program, denosumab demonstrated long-lasting reductions in bone turnover markers. Phase 2 studies demonstrated both increases in bone mineral density and reductions in bone turnover markers and were used to define the dose level to study in the Phase 3 program. The Phase 3 studies demonstrated robust reduction of fracture risk and, in 2010, led to the approval of denosumab as an agent to treat osteoporosis (Prolia®) in the United States.
It was hypothesized that RANKL is an important mediator of osteoclastic activity not only in osteoporosis but also in metastatic bone disease. Various rodent models of skeletal metastasis were found to have increases in stromal RANKL, and RANKL expression had been observed in some tumor types. Nonclinical studies conducted by Amgen and others established that RANKL inhibition can decrease osteoclast number and activity, limit the development of lytic bone lesions, and reduce skeletal tumor burden. Based on these positive findings from preclinical studies, a clinical development program was initiated and ultimately led, also in 2010, to the approval of denosumab (Xgeva®) for the prevention of skeletal-related events in patients with solid tumors.
Questions
1. The case describes an approach that first seeks to identify novel proteins, then determines whether the protein has a biologically meaningful role in a particular pathway(s), and finally develops a useful therapeutic that modulates the novel pathway. What are the advantages and disadvantages of such an approach compared to a program that starts with a validated biologic target? Which approach would you expect to be more likely to result in an approved therapeutic agent?
2. After the elucidation of the OPG–RANK–RANKL pathway, what types of protein constructs were engineered to modulate the pathway? What were the advantages and disadvantages of these different protein constructs?
3. What role did animal studies play in a discovery approach that was centered in genomics and bioinformatics?
4. What were the key data that would represent proof of concept that manipulation of the OPG–RANK–RANKL pathway could have beneficial effects in patients?

The term drug discovery refers to the process by which pharmaceutical, biotechnology, academic, and government laboratories identify or screen compounds to find potentially active therapeutic agents. The discussion below is focused on the discovery of low-molecular-weight, chemically synthesized compounds. Many of the described concepts and principles are similar for the discovery of biotherapeutics (monoclonal antibodies and related molecules, oligonucleotide-based molecules, and others) but, generally, many fewer such molecules are interrogated during the screening process. Screening consists of testing many compounds in assays relevant to the disease in question: a compound that passes such a screen is called a hit. If the compound or its structural derivatives continue to show promise after further biological and chemical characterization, it becomes a lead. Drug discovery should ideally be cost-effective, producing hits that have a high likelihood of conversion to leads and eventually to successful drugs (Fig. 51-1).
Two basic strategies are used to identify hits. In a compound-centered approach, a compound is identified by one of several methods (described below), and its biological profile is explored. If the compound displays desirable pharmacologic activity, it is refined and developed further. In a target-centered approach, which is now the more common mode, the putative drug target is identified first. The potential target could be a receptor thought to be involved in a disease process, a critical enzyme, or another biologically important molecule in the disease pathway. Once the target is identified, researchers search for compounds that interact with the target as agonists, antagonists, or modulators. The search may be systematic, using information about the structure of the target as a starting point (structural biology-based approach or simply structure-based approach), or it may take a shotgun approach, in which all the compounds in a large library of substances, synthesized via combinatorial chemistry, are tested in a high-speed automated assay. After any of these approaches identifies a hit, the hit is then often modified with the aid of specific knowledge about its target. For example, such knowledge can be used to design a high-throughput screen that will test the biologic activity of compounds generated by chemical modifications of the original hit.
Natural and Synthetic Compounds
Traditionally, drugs were discovered using a compound-centered approach. Many of the earliest drugs discovered were natural products isolated from plants, molds, or other organisms. Often, the discoveries were made serendipitously. For example, penicillin (see Chapter 35, Pharmacology of Bacterial and Mycobacterial Infections: Cell Wall Synthesis) was discovered when Alexander Fleming observed that spores of the contaminant mold Penicillium notatum inhibited bacterial growth in a petri dish. Other natural products that have been transformed into successful drugs include paclitaxel, a chemotherapeutic agent derived from the Pacific yew tree; morphine, an opioid analgesic obtained from the opium poppy, which has also been transformed into the drug oxycodone in a few synthetic steps; halichondrin, derived from the marine sponge Halichondria okadai, which was used as the lead for the synthesis of the anticancer drug eribulin (Halaven®); streptokinase, a thrombolytic agent obtained from streptococcal bacteria; and cyclosporine, an immunosuppressive agent obtained from a fungus. In recent years, there has also been a resurgence of interest in the use of cytotoxic natural products as “warheads” in antibody–drug conjugates (ADCs). Table 51-1 shows the structures of several drugs obtained from natural products.
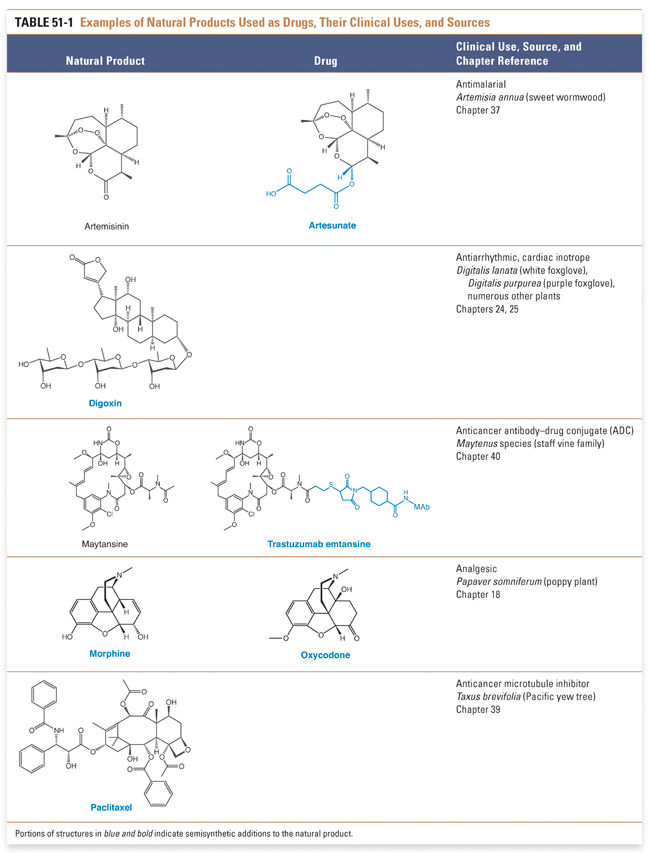
There are several advantages to examining natural products as a source for potential drugs. First, natural products have a reasonable likelihood of biological activity. Second, it may be easier to isolate a compound from its natural source than to synthesize a compound de novo, especially if the structure of the compound is complex or requires difficult synthetic manipulations. Paclitaxel, for example, has a complex structure that contains four fused rings, one of which contains eight carbons. A chemical synthesis of the compound took over 50 steps to complete and had a total yield of less than 1%. Third, it may be feasible to use the natural compound as a starting point for synthetic fine-tuning (i.e., to form a semisynthetic product). Natural products also have disadvantages: it often takes significant effort to isolate a natural product, without a guarantee of success, and, even if it is found to be pharmacologically active, a natural product can be expensive to isolate and modify. Also, since the in vivo mechanism of action is often unknown for many natural products, establishing screening systems and identifying clinically feasible safety monitoring tools for these highly potent compounds present significant challenges in the current environment of targeted pharmaceutical therapies.
Synthetic compounds are now frequently used to search for new drugs. Researchers can construct a library consisting of thousands of compounds with differing structural characteristics, tailored for a particular type of investigation. A library could, for example, consist of numerous compounds that have a phenylalanine–proline bond or that are likely agonists or antagonists of a particular class of receptors.
An alternative compound-centered approach uses the natural ligand (often an agonist) of a receptor as the starting point for drug development. For example, because lack of dopamine in the nigrostriatal pathways is associated with Parkinson’s disease (see Chapter 14, Pharmacology of Dopaminergic Neurotransmission), one of the first effective treatments involved administering the drug levodopa (L-DOPA), a metabolic precursor of dopamine. Insulin was developed in much the same way; once it was discovered that the signs and symptoms of diabetes were caused by low insulin levels, insulin was administered exogenously as an effective treatment.
The natural agonist for a receptor can also serve as a skeleton on which chemical modifications can be made. These changes can alter the compound’s binding affinity, physiologic effect (such as converting an agonist into an antagonist; see Chapter 1, Drug–Receptor Interactions), distribution, metabolism, or pharmacokinetics. This approach was employed in the development of cimetidine (see Chapter 44, Histamine Pharmacology), an H2 receptor antagonist. Starting with histamine, researchers made successive modifications in the structural skeleton to synthesize an antagonist with high affinity for the receptor and decreased toxicity. Similarly, modified insulins with different pharmacokinetic properties are now used to treat patients with diabetes.
Modifying a small-molecule agonist has a relatively high likelihood of success. Because the natural agonist is biologically active, chemical derivatives of that compound are also likely to be biologically active. Problems may also arise. Dopamine formed from exogenous L-DOPA can bind to receptors in areas of the brain other than the nigrostriatal pathways and cause hallucinations. Moreover, many disease processes are not mediated by the interaction of a small-molecule agonist and its receptor. Many targets for drug molecules, such as voltage-gated ion channels or intracellular signaling proteins, have no endogenous small-molecule agonists and hence are not amenable to the analogue approach.
In a target-centered approach to drug discovery and design, researchers use a biochemical or molecular target known to be essential in the disease of interest (a “validated” target) to search for hits. This approach has several advantages. First, if the target has been associated with a disease process, a hit that successfully interacts with the target has a relatively high likelihood of useful pharmacologic activity. Second, because the target is known, it may be easier to devise assays capable of isolating the effect of potential hits on the target. This is especially true for disease processes too complex to observe in cell or tissue preparations. For example, although a potential drug’s effect on the process of atherosclerosis may be difficult to measure rapidly, it is relatively easy to measure whether the drug inhibits an enzyme known to be involved in the pathogenesis of atherosclerosis, such as HMG-CoA reductase (see Chapter 20, Pharmacology of Cholesterol and Lipoprotein Metabolism). As knowledge of the pathophysiology of disease processes has increased, target-centered approaches to drug discovery have become increasingly successful, and most new drugs have been discovered using target-centered methods. HIV protease inhibitors, such as ritonavir, are notable examples of a small-molecule drug class discovered using a target-centered approach (see Chapter 38, Pharmacology of Viral Infections). In a complementary target-centered approach, dissection of the underlying biological pathway has allowed the development of macromolecules, including antibodies, as novel pharmaceuticals to interrupt the pathway (Box 51-1).
BOX 51-1 Macromolecular Biologics and Therapeutics |
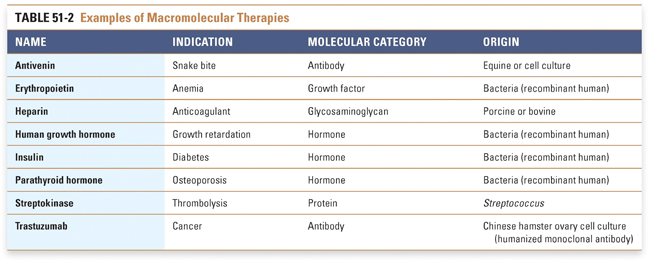
Increasingly, pharmaceutical and biotechnology companies are turning toward large molecules such as peptides, peptidomimetics, proteins, antisense oligonucleotides, and monoclonal antibodies. The pharmacologic properties and clinical utility of these therapies are described in Chapter 54, Protein Therapeutics. The approach to the discovery and development of these molecules can differ significantly from that for small molecules. Consider, for example, the development of agents for the treatment of diseases related to an insufficiency or lack of an endogenous compound, such as insulin for diabetes, erythropoietin for anemia, or a coagulation factor (factor VIII or factor IX) for an inherited coagulopathy. In these situations, referred to as replacement therapy, it is not necessary to perform extensive screening of a large number of molecules to determine whether the endogenous molecule needs to be modified. Therefore, these agents may rapidly move into development and human testing. Natural or modified macromolecules are increasingly used not only to replace but also to modulate physiologic processes, and engineered macromolecules such as antibodies are being used in the treatment of disease (Table 51-2). In the case of antibodies, the drug discovery and development process may involve modifications that increase the affinity or specificity of the antibody for the desired molecular target or that “humanize” the antibody in order to minimize its immunogenic potential. Because these types of molecules must typically be administered parenterally, the need to screen for acceptable pharmacokinetic properties is lessened. Furthermore, the required discovery biology and animal toxicity testing may not be as extensive because toxicities for biotherapeutics are generally related to “hyperpharmacology” and there is often less risk of “off-target” toxicity (see Chapter 6, Drug Toxicity). Manufacturing a biologic product is much more expensive and more technically challenging than manufacturing a chemically synthesized molecule. Major challenges in development of a biologic product are to develop a system capable of producing the desired macromolecule in a bacterium, yeast, or mammalian cell and then to isolate the compound in pure form from the large mixture of metabolic products that often result from the synthesis. Faithfully reproducing the complex procedures involved in macromolecule synthesis and purification makes the preparation of generic biologic drugs a substantial challenge. |

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


