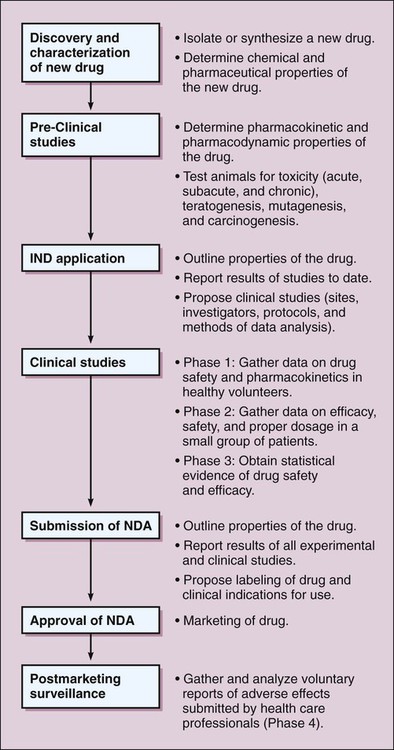
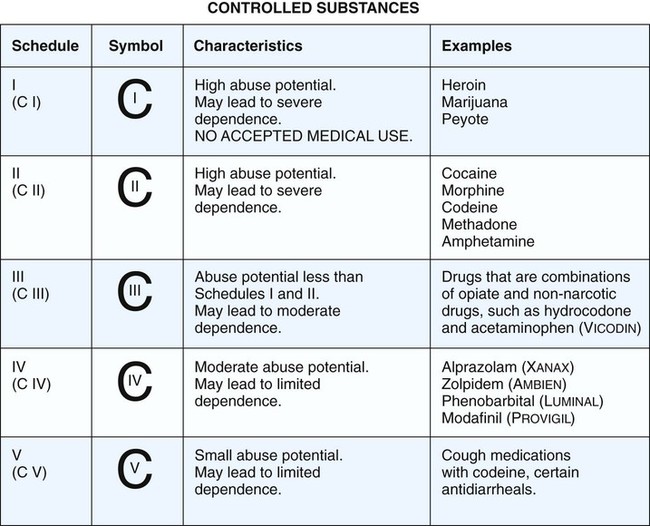
Drug development in most countries has many features in common, beginning with the discovery and characterization of a new drug and proceeding through the clinical investigations that ultimately lead to regulatory approval for marketing the drug. Steps in the process of drug development in the United States are depicted in Figure 4-1. Federal regulations require that extensive toxicity studies in animals be conducted to predict the risks that will be associated with administering the drug to healthy human subjects and patients. The value of the preclinical studies is based on the proven correlation between drug toxicity in animals and humans. As outlined in Table 4-1, the studies involve short-term and long-term administration of the drug and are designed to determine the risk of acute, subacute, and chronic toxicity, as well as the risk of teratogenesis, mutagenesis, and carcinogenesis. After animals are treated with the new drug, their behavior is assessed and their blood samples are analyzed for indications of tissue damage, metabolic abnormalities, and immunologic effects. Tissues are removed and examined for gross and microscopic pathologic changes. Offspring are also studied for adverse effects. TABLE 4-1 Drug Toxicity Studies in Animals *The LD50 (the median lethal dose) is the dose that kills half of the animals in a 14-day period after the dose is administered. The CSA classified drugs with abuse potential into five schedules, based on their degree of potential for abuse and their clinical usage (Fig. 4-2). Schedule I drugs are classified as having high abuse potential and no legitimate medical use, and their distribution and possession are prohibited. Schedule II drugs have high abuse potential but a legitimate medical use, and their distribution is highly controlled through requirements for inventories and records and through restrictions on prescriptions. Schedule III, IV, and V drugs have lower abuse potential and decreasingly fewer restrictions on distribution. The CSA requires that all manufacturers, distributors, physicians, and medical researchers using controlled drugs register with the Drug Enforcement Administration (DEA), which is responsible for enforcing the Act.
Drug Development and Safety
Drug Development
Preclinical Studies
TYPE OF STUDY
METHOD
OBSERVATIONS
Acute toxicity
Administer a single dose of the drug in two species via two routes.
Behavioral changes, LD50,* and mortality.
Subacute toxicity
Administer the drug for 90 days in two species via a route intended for humans.
Behavioral and physiologic changes, blood chemistry levels, and pathologic findings in tissue samples.
Chronic toxicity
Administer the drug for 6-24 months, depending on the type of drug.
Behavioral and physiologic changes, blood chemistry levels, and pathologic findings in tissue samples.
Teratogenesis
Administer the drug to pregnant rats and rabbits during organogenesis.
Anatomic defects and behavioral changes in offspring.
Mutagenesis
Perform the Ames test in bacteria. Examine cultured mammalian cells for chromosomal defects.
Evidence of chromosome breaks, gene mutations, chromatid exchange, trisomy, or other defects.
Carcinogenesis
Administer the drug to rats and mice for their entire lifetime.
Higher than normal rate of malignant neoplasms.
Federal Drug Laws and Regulations
Drug Abuse Prevention Laws
Harrison Narcotics Act
Comprehensive Drug Abuse Prevention and Control Act
Adverse Effects of Drugs
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


