Introduction
The pancreas is a gland with both exocrine and endocrine functions. The exocrine pancreas contains acini, which secrete pancreatic juice into the duodenum through the pancreatic ducts (Figure 15–1). Pancreatic juice contains a number of enzymes, some of which are initially secreted as zymogens in an inactive form. Once activated, these enzymes help to digest food and prepare it for absorption in the intestine. Disorders interfering with normal pancreatic enzyme activity (pancreatic insufficiency) cause maldigestion of fat and steatorrhea (fatty stools). Pathology of the exocrine pancreas results from inflammation (acute pancreatitis, chronic pancreatitis), neoplasm (ductal adenocarcinoma, neuroendocrine tumors, and other pancreatic neoplasms), or duct obstruction by stones or abnormally viscid mucus (cystic fibrosis).
The endocrine pancreas is composed of the islets of Langerhans. The islets are distributed throughout the pancreas and contain several different hormone-producing cells. The islet cells manufacture hormones such as insulin that are important in nutrient absorption, storage, and metabolism. Dysfunction of the endocrine pancreas may cause diabetes mellitus (see Chapter 18).
Both exocrine and endocrine pancreatic dysfunction occur together in some patients.
Normal Structure & Function of the Exocrine Pancreas
The pancreas is a solid organ that lies transversely in the retroperitoneum deep within the epigastrium. It is firmly fixed by fibrous attachments anterior to the suprarenal aorta and the first and second lumbar vertebrae. Thus, the pain of acute or chronic pancreatitis is situated deep in the epigastric region and frequently radiates to the back.
Normally, the pancreas is about 15 cm long, although the normal pancreas weighs less than 110 g. The organ is covered by a thin capsule of connective tissue that sends septa into it, separating it into lobules.
The pancreas can be divided into four parts: head, including the uncinate process; neck; body; and tail. The head is the thickest part of the gland (2–4 cm) and lies in the “C-loop” or curved space between the first, second, and third portions of the duodenum. The uncinate process is the portion of the head that extends dorsally and to the left behind the superior mesenteric vessels. The neck connects the head and body and sits immediately ventral to the superior mesenteric vessels. The body is situated transversely in the retroperitoneal space, bordered superiorly by the splenic artery and dorsally by the splenic vein. The tail of the pancreas is less fixed in the retroperitoneum and extends toward, and often immediately adjacent to, the hilum of the spleen.
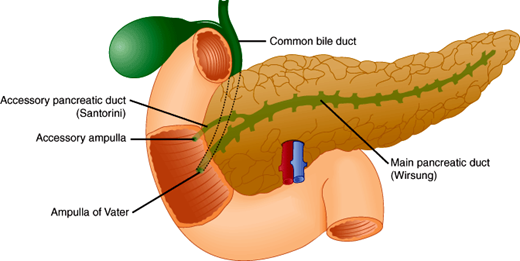
Embryologically, the pancreas develops as two separate endodermal buds from the developing foregut. These separate dorsal and ventral elements of the primordial pancreas initially develop opposite each other but with rotation of the primitive gut end up fusing together toward the left of the duodenum. The development of the dorsal and ventral buds is regulated by a complex process of intrinsic signals from the endodermal cells themselves, as well as extrinsic signals from surrounding mesoderm. The dorsal bud differentiates into the more cephalad and anterior portion of the pancreatic head, as well as the neck, body, and tail of the pancreas. The dorsal bud contains the accessory pancreatic duct (duct of Santorini), which enters the duodenum at the minor papilla. The smaller ventral bud arises adjacent to the bile duct to become the more caudal portion of the pancreatic head and the uncinate process. The ventral bud contains the major pancreatic duct (duct of Wirsung), which enters the duodenum at the ampulla of Vater (Figure 15–1) together with the common bile duct. As cellular and mesenchymal elements differentiate, the dorsal and ventral buds develop a conjoined ductal system, and the whole organ eventually takes its place in the retroperitoneum of the upper abdomen. Primitive neuroendocrine cells arise amid the developing ductal structures and eventually form the interspersed islets of Langerhans.
The major pancreatic duct of Wirsung normally becomes the drainage for the majority of the pancreatic parenchyma. This duct is normally about 3–4 mm in diameter. In most individuals, the pancreatic duct enters the duodenum at the duodenal papilla alongside the common bile duct, surrounded by the sphincter of Oddi, which controls the drainage of both ducts. In about one-third of individuals, the duct of Wirsung and the common bile duct join to form a common channel before terminating at the ampulla of Vater (Figure 15–1).
Pancreas divisum, the most common congenital anomaly of the pancreas, occurs when the embryologic ventral and dorsal components of the pancreas fail to fuse, thus leaving two distinct ductal systems that do not communicate and that separately drain into the duodenum through two different papillae. The smaller system drains through the major papilla, but the dominant dorsal system drains through the minor papilla. This situation can cause a relative obstruction to flow of pancreatic juice and may be associated with the development of pancreatitis. Pancreas divisum occurs in up to 7% of autopsy series.
The exocrine pancreas consists of clusters of enzyme-secreting acini centered around and individually drained by ductules. The islets of Langerhans of the endocrine pancreas are clusters of a few hundred hormone-secreting cells, each located between the lobules.
Each pancreatic acinus is composed of several acinar cells surrounding a lumen (Figure 15–2). Centroacinar cells are centrally located in the acini, interpositioned between the acinar cells and ductal epithelium. The centroacinar cells are believed to have a primary role in the secretion of electrolytes and water into the pancreatic ductal system. The acinar cells synthesize and secrete enzymes. On histologic examination, acinar cells are typical exocrine glandular cells. They are pyramidal epithelial cells arranged in rows. Their apexes join to form the lumen of the acinus. Zymogen granules containing digestive enzymes or their precursors are found in the acinar cells. These granules are discharged by exocytosis from the apexes of the cells into the lumen. The number of zymogen granules in the cells varies; more are found during fasting and fewer after a meal.
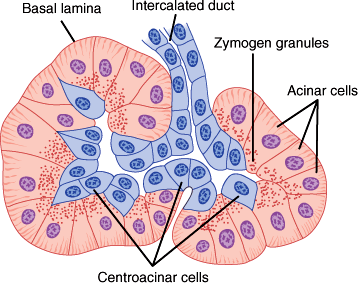
Acini are centered on small branches of the pancreatic duct, which eventually converge to form the continuous lumen of the main pancreatic duct. The mature ducts are lined with a continuous layer of ductal epithelial cells, joined by tight junctions. The ductal epithelium contributes to the secretion of water and electrolytes into pancreatic secretions and forms the important epithelial barrier separating the pancreatic parenchyma from the enzyme-rich ductal secretions. Compromise of this epithelial barrier due to inflammation or trauma may be associated with significant peripancreatic inflammation and severe clinical sequelae.
As much as 1500 mL of pancreatic juice is secreted each day by a normal pancreas. Disease states (eg, chronic pancreatitis) may be associated with a marked decrease in exocrine pancreatic secretion. Pancreatic juice contains water, ions, and a variety of proteins. The principal ions in pancreatic juice are HCO3−, Cl−, Na+, and K+. Of these, HCO3− is particularly important. At maximum flow rates, the concentration of HCO3− in pancreatic juice may reach 150 mEq/L (vs. 24 mEq/L in plasma), and the pH of the juice may reach 8.3. The alkaline nature of pancreatic juice plays a major role in neutralizing the gastric acid entering the duodenum with ingested food (chyme) from the stomach. The pH of the duodenal contents rises to 6.0–7.0, and by the time the chyme reaches the jejunum, its pH is nearly neutral.
Proteomic analysis suggests that more than 200 proteins exist in pancreatic secretions. Many of these ubiquitous proteins have various roles in cellular growth and signaling, while others are engaged in cellular immunology. The remainder of the pancreatic proteins secreted are responsible for the digestive functions of the exocrine pancreas. A primary function of the pancreas is the digestion of proteins in partially digested ingested food. This digestion is mediated by trypsinogen and other secreted proteases. However, the exocrine pancreas also secretes enzymes responsible for metabolism and absorption of lipids (lipase, colipase) and carbohydrates (amylase, enolase).
Some of the pancreatic enzymes (lipase, amylase, deoxyribonuclease, and ribonuclease) are secreted by the acinar cells in their active forms. The remaining enzymes are secreted as inactive proenzymes or zymogens (trypsinogen, chymotrypsinogen, proelastase, procarboxypeptidase, and phospholipase A2) that are activated in the lumen of the proximal intestine. Aberrant activation of zymogens within the acinar cell is hypothesized to lead to acute pancreatitis and pancreatic autodigestion.
When the pancreatic juice enters the duodenum, trypsinogen is converted to the active form trypsin by an enzyme found in the intestinal brush border called enteropeptidase. Trypsin then converts the remaining proenzymes into active enzymes (eg, chymotrypsinogen into chymotrypsin). Trypsin can also activate its own precursor, trypsinogen, producing the potential for an autocatalytic chain reaction.
When trypsinogen is activated within the pancreas itself, two known protective mechanisms are available. First, there is inhibition of activated trypsin by pancreatic secretory trypsin inhibitor (PSTI), also known as serine protease inhibitor, Kazal type 1 (or SPINK1), which can inhibit approximately 20% of trypsin activity. If trypsin activity overwhelms the SPINK1/PSTI inhibitory capacity, trypsin inactivation can occur through trypsin autolysis.
Recent advances in our understanding of pancreatic exocrine function reveal hormonal and neural factors as two distinct but interactive elements that regulate secretion. Two hormones in particular appear to have a primary role in the secretion of pancreatic enzymes, secretin and cholecystokinin (CCK). Both hormones are produced by specialized enteroendocrine cells of the duodenal mucosa and act by distinct but synergistic intracellular pathways on the pancreatic acinar cells.
Secretion of secretin is triggered by gastric acid and by the products of protein digestion in the duodenum. Secretin acts chiefly on the pancreatic ductal epithelial, centroacinar, and to a lesser extent acinar cells to produce HCO3−, thus raising the pH of the pancreatic secretions. Secretion of H2O is also increased in response to secretin, increasing the absolute volume of pancreatic juice. Mechanistic studies have demonstrated that secretin and the related hormone vasoactive intestinal peptide (VIP) act on ductal and acinar cells by activation of adenylate cyclase and subsequent cAMP-dependent protein kinase A. Proteomic analysis has revealed that secretin does not appear to alter the constituents of pancreatic juice but instead regulates the relative proportions of secreted enzymes.
Secretion of CCK is triggered by the products of protein and fat digestion (peptides, amino acids, and fatty acids) when they enter the duodenum. The release of CCK from specific intestinal cells appears to be regulated by a cholecystokinin-releasing peptide in the proximal small intestine that is trypsin sensitive and active in the lumen. CCK controls pancreatic exocrine secretion through two mechanisms: 1) activation of neurons located in the dorsal motor nucleus of the vagus motor neurons that control parasympathetic signals, and 2) direct action on pancreatic acinar cells. CCK release raises intracellular Ca2+ concentrations, which lead to release of pancreatic enzymes from the zymogen granules. Related enteric hormones acetylcholine and gastrin-releasing peptide (GRP) appear to act by similar calcium-dependent pathways. The integrated action of both secretin and CCK produces abundant secretion of enzyme-rich, alkaline pancreatic juice.
When both cAMP and calcium-dependent pathways are stimulated, the effect within the acinar cell is greater than the sum of their individual activities. Thus, CCK and secretin appear to act synergistically in response to a meal to stimulate the production of a large volume of alkaline pancreatic juice rich in digestive enzymes. Recent evidence also implicates a number of other gastrointestinal (GI) hormones and peptides (ghrelin, leptin, and melatonin) in the regulation of pancreatic endocrine and exocrine secretion.
The secretion of pancreatic juice aids digestion in several ways. The large amount of bicarbonate in the juice helps to neutralize the acidic chyme from the stomach so that the pancreatic enzymes can function optimally in a neutral pH range.
Each of the enzymes also has an important digestive function. In digesting carbohydrates, pancreatic amylase splits straight-chain glucose polysaccharides (so-called amyloses in starch) into smaller α-limit dextrins, maltose and maltotriose. Brush border enzymes in the small intestine complete the hydrolysis of these smaller sugars into glucose, which is transported across the intestinal epithelium by Na+-coupled transport. Pancreatic lipase contributes to fat metabolism by hydrolyzing triglycerides into fatty acids and a monoglyceride; this activity is most efficient in the presence of bile acids, which serve to emulsify the triglycerides. Phospholipase A2 splits a fatty acid off from lecithin to form lysolecithin. Ribonuclease and deoxyribonuclease attack the nucleic acids. The remaining enzymes help to digest proteins. Trypsin, chymotrypsin, and elastase are endopeptidases (ie, they cleave peptide bonds in the middle of polypeptide chains). Carboxypeptidase is an exopeptidase (ie, it splits peptide bonds adjacent to the carboxyl terminals of peptide chains). Together, these proteases break down proteins into oligopeptides and free amino acids.
Checkpoint
1. What histologic features are associated with pancreatic secretion of digestive enzymes into the GI tract?
2. What is the volume, composition, and function of pancreatic juice?
3. What are the neural and hormonal controls over exocrine pancreatic function?
4. Why does trypsinogen not self-activate before arriving in the duodenum?
Pathophysiology of Selected Exocrine Pancreatic Disorders
Acute pancreatitis is a clinical syndrome resulting from acute inflammation and destructive autodigestion of the pancreas and peripancreatic tissues. Acute pancreatitis is the third most common indication for hospital admission among GI diseases and is associated with significant morbidity and mortality. Data from the National Center for Health Statistics clearly document a near doubling of cases of hospital admissions due to acute pancreatitis between 1985 and 2005. Fortunately, the overall survival rate of patients with acute pancreatitis is increasing, despite the continued high mortality rate (20–25%) in patients admitted to the intensive care unit with severe acute pancreatitis.
Acute pancreatitis has many causes, as summarized in Table 15–1. In clinical practice, biliary tract disease and alcohol ingestion account for the majority of cases, with metabolic causes, mechanical etiologies, drug reactions, and traumatic injuries accounting for almost all of the remaining cases. Regardless of etiology, the pathogenesis of pancreatic injury, associated systemic effects, and risk factors for severe acute pancreatitis appear to be similar.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alcohol use is commonly associated with acute pancreatitis in developed countries. Acute pancreatitis typically occurs after a binge of heavy drinking; chronic heavy alcohol ingestion clearly may lead to chronic pancreatitis and may increase susceptibility to episodes of acute pancreatitis. A number of mechanisms are responsible for alcohol-induced damage to the pancreas. Alcohol or its metabolite, acetaldehyde, can have a direct toxic effect on pancreatic acinar cells, leading to intracellular trypsin activation by the lysosomal enzymes. Additionally, inflammation of the sphincter of Oddi can lead to retention of hydrolytic enzymes in the pancreatic duct and acini. Malnutrition may predispose alcoholics to pancreatic injury. For example, deficiencies of trace elements such as zinc or selenium occur in alcoholic patients and are associated with acinar cell injury. Metalloenzymes such as superoxide dismutase, catalase, and glutathione peroxidase are important scavengers of free radicals.
In patients who do not drink alcohol, the most common cause of acute pancreatitis is biliary tract disease. In such cases, the hypothesized mechanism is obstruction of the common bile duct and the main pancreatic duct when a gallstone or biliary sludge becomes lodged at the ampulla of Vater. Reflux of bile or pancreatic secretions into the pancreatic duct leads to parenchymal injury. Others have proposed that bacterial toxins or free bile acids travel via lymphatics from the gallbladder to the pancreas, giving rise to inflammation. In either case, acute pancreatitis associated with biliary tract disease is more common in women because gallstones are more common in women.
A significant proportion of “gallstone” pancreatitis is not associated with discrete, measurable gallstones passing through the bile duct and obstructing the ampulla. Instead, biliary sludge, or microlithiasis, is believed to play an etiologic role in many cases of pancreatitis, which were previously classified as idiopathic. Endoscopic retrograde cholangiopancreatography (ERCP) performed in such cases often identifies the microlithiasis and viscous particulate bile in the distal common bile duct, which can cause transient biliary obstruction and activate the same mechanistic pathways that lead to pancreatitis as happens with larger gallstones. An alternative mechanism that has been proposed is recurrent passage of microlithiasis causing papillary stenosis or sphincter of Oddi dysfunction.
Thus, the absence of obvious gallstones on imaging studies does not definitively rule out a biliary cause of acute pancreatitis. Biliary microlithiasis may be suspected when an ultrasound shows low-level echoes that gravitate toward the dependent portion of the gallbladder without the acoustic shadowing typical of gallstones. Microlithiasis is documented when cholesterol monohydrate crystals and calcium bilirubinate granules are found on light microscopy of an endoscopically acquired, centrifuged specimen of bile. In clinical practice, this diagnosis is often made in a patient with an appropriate presentation and with risk factors for biliary microlithiasis including pregnancy, rapid weight loss, critical illness, prolonged fasting, total parenteral nutrition, administration of certain drugs (ceftriaxone and octreotide), and bone marrow or solid organ transplantation.
Acute pancreatitis may result from a variety of infectious agents, including viruses (mumps virus, coxsackievirus, hepatitis A virus, HIV, or cytomegalovirus) and bacteria (Salmonella typhi or hemolytic streptococci). Patients with HIV infection can develop acute pancreatitis from the HIV infection itself, from related opportunistic infections, or from antiretroviral therapies. In HIV-infected patients, pancreatitis has been associated with intravenous drug abuse, pentamidine therapy, Pneumocystis jirovecii and Mycobacterium avium-intracellulare infections, and gallstones.
Blunt or penetrating trauma and other injuries may cause acute pancreatitis. Pancreatitis sometimes occurs after surgical procedures near the pancreas (duodenal stump syndrome, pancreatic tail syndrome after splenectomy). Shock and hypothermia may cause decreased perfusion, resulting in cellular degeneration and release of pancreatic enzymes. Radiation therapy of retroperitoneal malignant neoplasms can sometimes cause acute pancreatitis, likely by injury to the microvasculature and acinar architecture.
Marked hypercalcemia, such as that associated with hyperparathyroidism, sarcoidosis, hypervitaminosis D, or multiple myeloma, causes acute pancreatitis in about 10% of cases. Two mechanisms have been hypothesized. The high plasma calcium concentration may cause calcium to precipitate in the pancreatic duct, leading to ductal obstruction. Alternatively, hypercalcemia may stimulate activation of trypsinogen in the pancreatic duct.
Pancreatitis is also associated with hyperlipidemia, particularly those types characterized by increased plasma levels of chylomicrons (types I, IV, and V). In these cases, it is postulated that free fatty acids liberated by the action of pancreatic lipase cause gland inflammation and injury. Alcohol abuse or oral contraceptive use increases the risk of acute pancreatitis in patients with hyperlipidemia.
A variety of drugs have been associated with pancreatitis, including corticosteroids, thiazide diuretics, immunosuppressants, and cancer chemotherapeutic agents.
Rarely, acute pancreatitis may be familial, occurring with an autosomal dominant inheritance pattern. Hereditary pancreatitis typically presents as recurring acute pancreatitis in childhood, progressing to chronic pancreatitis by young adulthood in more than 50% of cases. Hereditary recurrent acute pancreatitis has been associated with mutations of the cationic trypsinogen gene (protease, serine, 1; PRSS1) mapped to chromosome 7q35. Two point mutations, R122H and N29I, account for most cases and can be detected by genetic testing. Studies have suggested that the R122H mutation is associated with more severe acute pancreatitis, leading to more frequent attacks and hospital admissions. Other families have mutations in SPINK1/PSTI. It appears that mutations in cationic trypsinogen enhance trypsinogen autoactivation by altering calcium-mediated regulatory pathways, and mutations in SPINK1/PSTI diminish inhibition of active trypsinogen. Other mutations eliminate the trypsin autolysis site. Patients demonstrated to have hereditary pancreatitis should be enrolled in a pancreatic cancer surveillance program, and total pancreatectomy should be considered in select cases, as approximately 40% of affected patients develop pancreatic cancer by age 70 years.
In recent years, our understanding of the diagnosis and classification of autoimmune pancreatitis has evolved. This chronic disease of fibrosis and lymphoplasmacytic inflammation may cause both acute episodes of pancreatitis as well as chronic injury. Two subtypes have been characterized. Type I autoimmune pancreatitis accounts for more that 80% of cases in the United States and is associated with elevated serum levels of IgG4 and with lymphocytic infiltration throughout the pancreatic parenchyma. Many patients with type I autoimmune pancreatitis have extrapancreatic manifestations and are often classified as having IgG4-related disease. Type II autoimmune pancreatitis is more common outside of the United States and does not appear to be IgG4-mediated. The pathognomonic histopathologic findings in this disease are granulocyte-epithelial lesions with neutrophilic infiltration. Type II autoimmune pancreatitis more often presents with acute pancreatitis compared with type I disease.
In about 15–25% of cases of acute pancreatitis, no etiologic factor can be identified. Idiopathic acute recurrent pancreatitis is seen in patients with more than one attack of acute pancreatitis when the underlying cause eludes detection despite a thorough search.
The symptoms, signs, laboratory findings, and complications of acute pancreatitis can be explained on the basis of the pathologic damage to the ductules, acini, and islets of the pancreas. However, both the degree of damage and the clinical consequences are quite variable.
When the damage is limited in extent, the pathologic features consist of mild to marked swelling of the gland, especially the acini, and mild to marked infiltration with polymorphonuclear neutrophils. However, damage to tissue is usually only minimal to moderate, and there is no hemorrhage. In some cases, suppuration may be found along with edema, and this may result in tissue necrosis and abscess formation. In severe cases, massive necrosis and liquefaction of the pancreas occur, predisposing to pancreatic abscess formation. Vascular necrosis and disruption may occur, resulting in peripancreatic hemorrhage. While microvascular hemorrhage involving peripancreatic tissue is common in severe cases of acute pancreatitis, significant bleeding from large vessel erosion is a rare clinical entity and is more often seen in chronic pancreatitis.
Severe cases of pancreatitis may be associated with the formation of ascites, which is likely a combination of serous fluid excreted by the inflamed peritoneal surface, liquefied peripancreatic fat, blood from peripancreatic tissues, and necrotic pancreatic debris. In rare cases associated with ductal disruption of the pancreas, the ascites may contain frank pancreatic secretions rich in amylase and other pancreatic enzymes. Documentation of amylase-rich peritoneal fluid establishes the diagnosis of so-called pancreatic ascites. In cases of severe acute pancreatitis, the peritoneal surfaces have a characteristic appearance upon surgical exploration or autopsy; fat necrosis, or saponification, may occur in and around the pancreas, omentum, and mesentery, appearing as chalky white foci that may later calcify.
Histologic studies of pancreas tissue obtained from patients with first attacks of acute alcoholic pancreatitis who underwent surgery for complications have found that the acute pancreatitis (pancreatic necrosis, steatonecrosis, infiltration by inflammatory cells) sometimes develops in a gland already affected by chronic pancreatitis (perilobular and intralobular fibrosis, loss of exocrine parenchyma and atrophy of residual lobules, dilated interlobular and intralobular ducts lined with cuboidal or flattened epithelium, and protein plugs within dilated ducts). It has been conjectured that, if acute alcoholic pancreatitis develops in a pancreas already affected by chronic pancreatitis, it is due to obstruction of the ducts by protein plugs, an early lesion of chronic pancreatitis.
The pathogenesis of acute pancreatitis remains only partially understood. The central theory in this disease has long centered on the aberrant activation of trypsinogen and other enzymes within the pancreatic acini, causing autodigestion and a profound systemic inflammatory response. Recent evidence suggests that other events parallel to trypsinogen activation occur, such as the activation of NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells, a protein complex that controls the transcription of DNA) that can induce acute pancreatitis in experimental models (Figure 15–3). However, elegant studies have confirmed that expression of active trypsin within pancreatic acini is itself sufficient to induce cell death and inflammation in acute pancreatitis. Thus, the in vivo role of alternative mechanisms of pancreatic autodigestion remains unclear.
Figure 15–3
This schematic shows parallel cell-signaling pathways producing the pathologic effects, presumed to be trypsinogen and NFκB activation, leading to pancreatitis. At the bottom, caerulein (a CCK analog) binds to its receptor CCKA (cholecystokinin receptor subtype A) and leads, via Gq (G-protein q subtype) and phospholipase C (PLC), both to generation of inositol-3 phosphate (IP3) from phosphoinositol 4-phosphate (PIP2) and to generation of diacylglycerol (DAG). On the left, IP3 opens its endoplasmic reticulum (ER) membrane receptors, receptors that are implicated in physiologic calcium signaling. The calcium thus released leads caerulein to the pathologic effects inducing pancreatitis. On the right, DAG stimulates the release of two forms of protein kinase C (PKC), which in turn leads to the generation of protein kinase D subtype 1 (PKD1) and the pathologic effects inducing pancreatitis. (Redrawn, with permission, from Sah RP et al. Molecular mechanisms of pancreatic injury. Curr Opin Gastroenterol. 2011 Sept; 27(5):448.)
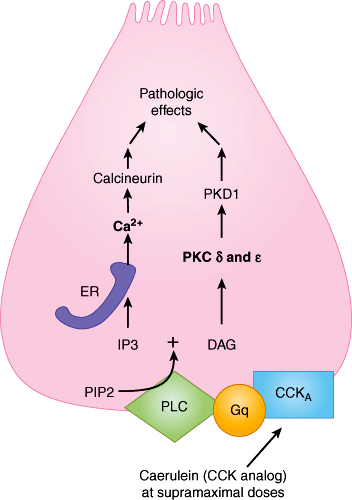
Typsinogen activation is associated with a sustained cytosolic influx of calcium (Ca2+) mediated by calcium channels in the plasma membrane as well as by calcium receptors in the endoplasmic reticulum. Calcineurin is a likely downstream target of elevated intracellular Ca2+ levels, mediating some of the injury observed in acute pancreatitis via T-cell activation.
Trypsinogen is likely activated within membrane-bound intracellular compartments that exhibit dysregulated autophagy in the setting of acute pancreatitis. While cathepsin B within lysosomes has been shown to activate trypsinogen, this likely occurs only in certain pathologic conditions such as a low intracellular pH. The mechanism of pH disturbance within acinar cells is likely due to an alteration in cell signaling mechanisms and inhibition of acinar bicarbonate secretion. Moreover, although cathepsin L (an alternate isoform of cathepsin B) normally degrades trypsin in an important cellular protective mechanism, disturbance of the intracellular environment has been shown to contribute to an imbalance in cathepsin B activity relative to cathepsin L activity.
The pathogenesis of alcoholic pancreatitis may be unique and may involve disordered agonist-receptor interaction on the membrane of pancreatic acinar cells. According to this theory, alcohol increases activation of intrapancreatic digestive enzymes either by sensitizing acinar cells to pathologic stimuli or by stimulating the release of the secretagogue, cholecystokinin (CCK), from duodenal cells. The hyperstimulation of pancreatic acinar cells and their muscarinic receptors mimics the mechanism of acute pancreatitis caused by scorpion stings, anti-acetylcholinesterase-containing insecticide poisoning, or administration of supramaximal doses of secretagogues such as acetylcholine and CCK. CCK receptor activation can initiate different patterns of zymogen activation in pancreatic acinar cells, and the extent of activation is enhanced by a distinct set of short-chain alcohols. Whether ethanol or other alcohols mediate these effects by interfering with acinar cell signaling pathways or by affecting acinar cell membrane fluidity is currently under investigation.
The pathologic changes result from the action of activated trypsin and other pancreatic enzymes on the pancreas and surrounding tissues. Activated trypsin in turn activates the proenzymes of chymotrypsin, elastase, and phospholipase A2, and those enzymes cause damage in several ways (Figure 15–4). For example, chymotrypsin activation leads to edema and vascular damage. Similarly, elastase, once activated from proelastase, digests the elastin in blood vessel walls and causes vascular injury and hemorrhage; damage to peripancreatic blood vessels can lead to hemorrhagic pancreatitis. Phospholipase A2 splits a fatty acid off lecithin, forming lysolecithin, which is cytotoxic to erythrocytes and damages cell membranes. Formation of lysolecithin from the lecithin in bile may contribute to disruption of the pancreas and necrosis of surrounding fat. Phospholipase A2 also liberates arachidonic acid, which is then converted to prostaglandins, leukotrienes, and other mediators of inflammation, contributing to coagulation necrosis. Pancreatic lipase, released as a direct result of pancreatic acinar cell damage, acts enzymatically on surrounding adipose tissue, causing the characteristic peripancreatic fat necrosis seen in severe acute pancreatitis (Figure 15–4).
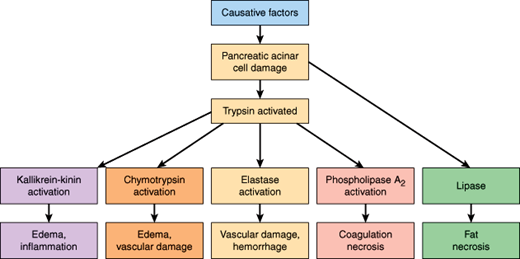
Furthermore, trypsin and chymotrypsin activate kinins, complement, coagulation factors, and plasmin, leading to edema, inflammation, thrombosis, and hemorrhage within the gland. For example, trypsin activation of the kallikrein-kinin system leads to the release of bradykinin and kallidin, causing vasodilation, increased vascular permeability, edema, and inflammation (Figure 15–4), which all contribute to the systemic inflammatory response syndrome characteristic of acute pancreatitis. Circulating phospholipases interfere with the normal function of pulmonary surfactant, contributing to the development of an adult respiratory distress syndrome in some patients with acute pancreatitis. Elevated serum lipase levels are sometimes associated with fat necrosis outside of the abdomen.
Experimental models of acute pancreatitis suggest that NFκB activation occurs in parallel with trypsinogen activation. Pathologic Ca2+ influx appears to play a role in NFκB activation and may be a common activator of both parallel pathways of pancreatic injury. Moreover, as a downstream effector of CCK action, protein kinase C isoforms appear to play a role in NFκB activation, consistent with parallel activation of zymogen and NFκB pathways.
Finally, during acute pancreatitis, both the CC and CXC families of cytokines are implicated in the pathogenesis of the local and systemic inflammatory response. Cytokines and other inflammatory mediators such as tumor necrosis factor (TNF), interleukins (especially IL-1, IL-6, and IL-8), platelet-activating factor (PAF), and endotoxin are released rapidly and predictably from inflammatory cells. This release appears to be in response to the presence of active digestive enzymes, independent of the underlying cause. Production of cytokines during clinical pancreatitis begins shortly after pain onset and peaks 36–48 hours later. These agents are now thought to be principal mediators in the transformation of acute pancreatitis from a local inflammatory process to a systemic illness (Figure 15–5). The degree of TNF-induced inflammation correlates with the severity of pancreatitis. Cytokines rapidly enter the systemic circulation from the peritoneal cavity via the thoracic duct. In the systemic circulation, the cytokines affect many body systems and can produce the systemic inflammatory response syndrome (SIRS) and the multiorgan dysfunction syndrome typical of severe acute pancreatitis. Systemic complications of acute pancreatitis, such as respiratory failure, shock, and even multisystem organ failure, are accompanied by significant increases in monocyte secretion of TNF, IL-1, IL-6, and IL-8, and upregulation of the number of receptors for these cytokines on target cells. This finding suggests that TNF, IL-1, IL-6, and IL-8 play a central role in the pathophysiology of these manifestations.
Figure 15–5
Inflammatory mediators of acute pancreatitis include interleukin-1B (IL-1) and tumor necrosis factor (TNF). As depicted, these two cytokines can induce other inflammatory mediators, such as IL-2, IL-6, IL-8, and IL-10; nitric oxide (NO); platelet activating factor (PAF); and interferon (INF)-α and INF-γ, while at the same time producing a direct noxious effect on the pancreas itself. Each of the mediators shown plays a role in development of the systemic manifestations of acute pancreatitis. ARDS, acute respiratory distress syndrome; ATN, acute tubular necrosis. (Redrawn, with permission, from Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg. 1998;175:76.)
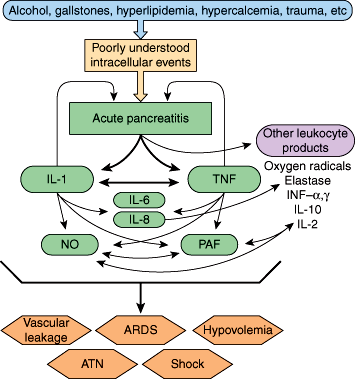
Studies also suggest that substance P acting via neurokinin-1 (NK-1) receptors, PAF, and chemokines interacting with CCR1 receptors play important proinflammatory roles in determining the severity of acute pancreatitis. In particular, substance P and neurokinin-1 are involved in mediating acute lung injury. Substance P, a neuropeptide released from sensory afferent nerve endings, binds to the NK-1 receptor on the surface of effector cells and increases the permeability of vascular endothelium. The amount of substance P in the pancreas is increased during episodes of acute pancreatitis, and acinar cell expression of NK-1 receptors is markedly upregulated. Substance P appears to be a powerful proinflammatory mediator of both pancreatitis and associated lung injury. PAF also appears to play an important role in the development of pancreatitis and associated lung injury. Chemokines are chemoattractant cytokines that are involved in the activation and trafficking of various inflammatory cells. Chemokines acting via the chemokine receptor CCR1 appear to have a role in determining the severity of pancreatitis-associated lung injury but no effect on the severity of the pancreatitis itself. On the other hand, complement factor 5a (C5a) appears to act as an anti-inflammatory agent during the development of pancreatitis.
Various factors play active roles as proinflammatory or anti-inflammatory agents in acute pancreatitis. Drugs or other interventions to counteract those agents that are proinflammatory (eg, TNF, IL-1, IL-6, IL-8, and PAF) or to stimulate those that are anti-inflammatory (eg, IL-10) may eventually prove useful in treating patients with clinical pancreatitis to prevent severe injury to the pancreas and to prevent associated systemic manifestations, such as lung injury.
Acute pancreatitis may present in a highly variable manner, with the severity of inflammation and associated morbidity differing markedly among patients. Approximately 80% of patients experience a mild, self-limited illness to 2–3 days with no significant sequelae, but the remainder may develop severe acute pancreatitis, a life-threatening illness defined by the presence of associated organ system failure (typically pulmonary, cardiovascular, and/or renal systems). Acute pancreatitis may recur, depending primarily on its cause. With repeated attacks, the gland may eventually be permanently damaged, resulting in chronic pancreatitis or sometimes pancreatic insufficiency (see below). The distinction between acute pancreatitis and an acute exacerbation of chronic pancreatitis is determined by the clinical history and the characteristic finding on imaging of chronic pancreatitis. Acute and chronic pancreatitis have notably different management paradigms, so this distinction is important.
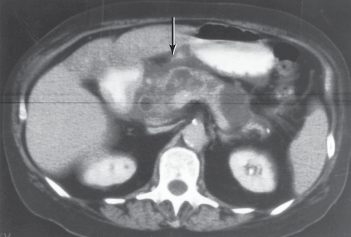
Recent consensus criteria require two of the following three criteria for the diagnosis of acute pancreatitis: abdominal pain, elevation of serum amylase or lipase (>3 times the upper limit of normal), and characteristic findings on computed tomography (CT) (or magnetic resonance imaging [MRI] or ultrasound). In practice, the first two elements are often present and suffice to make to the clinical diagnosis. Nonetheless, cross-sectional imaging (eg, contrast-enhanced CT may be useful in severe acute pancreatitis to assess the extent of associated pancreatic necrosis and other associated complications of the disease (Figure 15–6).
Abdominal pain is nearly universal and a hallmark presentation of acute pancreatitis. In rare cases, patients may present with occult pancreatic inflammation evident by hyperamylasemia—eg, following pancreatic trauma, medication administration, or other known precipitants. However, such presentations are unlikely to be associated with clinically significant pancreatitis.
The pain of acute pancreatitis is characteristic, often described as an intense, deep, searing pain that radiates to the back. Frank peritoneal inflammation may lead to diagnostic confusion with other, more immediate surgical emergencies such as a perforated peptic ulcer, appendicitis, or diverticulitis.
The pain of acute pancreatitis is thought to derive in part from stretching of the pancreatic capsule by distended ductules and parenchymal edema, inflammatory exudate, digested proteins and lipids, and hemorrhage. In addition, these materials may seep out of the parenchyma into the retroperitoneum and lesser sac, where they irritate retroperitoneal and peritoneal sensory nerve endings and produce intense back and flank pain. The clinical findings of generalized peritonitis may follow.
Stretching of the pancreatic capsule may also produce nausea and vomiting. Increasing abdominal pain, peritoneal irritation, and electrolyte imbalance (especially hypokalemia) may cause a paralytic ileus with marked abdominal distention. If gastric motility is inhibited and the gastroesophageal sphincter is relaxed, there may be emesis. Both small and large bowel often dilate during an acute attack. Sometimes only a localized segment of bowel dilates. For example, there may be localized dilation of a segment of jejunum overlying the pancreas. In such cases, a plain x-ray film of the abdomen shows thickening of the valvulae conniventes and air-fluid levels (“sentinel loop”). In other cases, there may be segmental dilation of a portion of the overlying transverse colon. The x-ray film shows a sharply demarcated area of localized colonic dilation and edema (“colon cutoff sign”).
Almost two-thirds of patients with acute pancreatitis develop fever. The pathophysiologic mechanism responsible for fever involves the extensive tissue injury, inflammation, and necrosis and release of endogenous pyrogens, principally IL-1, from polymorphonuclear leukocytes into the circulation. In most cases of acute pancreatitis, fever does not indicate a bacterial infection. However, persistent fever beyond the fourth or fifth day of illness—or spiking temperatures to 40°C or more—may signify development of infectious complications such as infected peripancreatic fluid collections, infected pancreatic necrosis, or ascending cholangitis.
The cardinal laboratory finding in acute pancreatitis is elevation of the serum amylase, often up to 10- to 20-fold. The serum amylase elevation occurs almost immediately (within hours), but it usually returns to normal within 48–72 hours even if symptoms continue. The sensitivity of the serum amylase in acute pancreatitis is estimated to be 70–95%, meaning that 5–30% of patients with acute pancreatitis have normal or minimally elevated serum amylase values. The specificity of the test is considerably lower. Patients with marked (more than 3-fold) elevations of serum amylase usually have acute pancreatitis. Patients with lesser elevations of serum amylase often have one of a variety of other conditions.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


