Chapter 18
Disorders of the Central and Peripheral Nervous Systems and the Neuromuscular Junction
Barbara J. Boss and Sue E. Huether

Central Nervous System Disorders
Traumatic Brain and Spinal Cord Injury
Traumatic Brain Injury
Traumatic brain injury (TBI) is an alteration in brain function or other evidence of brain pathology caused by an external force.1 An estimated 1.7 million people sustain TBI in the United States each year with approximately 53,000 deaths. Those at highest risk for TBI are children 4 years and younger, adolescents 15 to 19 years, and adults 65 years and older. Males have the highest incidence in every age group.2 TBI is highest among American Indian/Alaska Natives and blacks and in lower- and median-income families. TBIs are caused by falls (35%), motor vehicle–related injuries (17%), and a strike or blow to the head from or against an object (e.g., workplace or sports-related injuries [16.5%], assaults [10%], and other and unknown causes [21%]).3
Traumatic brain injury can be caused by closed (blunt) trauma and open (penetrating) trauma. Closed (blunt) trauma is more common and involves either the head striking a hard surface or a rapidly moving object striking the head. The dura mater remains intact, and brain tissues are not exposed to the environment. Most closed (blunt) trauma is mild (75% to 90%) and causes mild concussion and classic cerebral concussion (see pp. 587-588 and Table 18-1). Open (penetrating) trauma occurs when a break in (penetration of) the dura mater results in exposure of the cranial contents to the environment.
TABLE 18-1
SEVERITY OF BLUNT TRAUMA, TRAUMA STATE INDUCED, AND ONSET AND PERSISTENCE OF CLINICAL MANIFESTATIONS
| SEVERITY OF TRAUMA | TRAUMA STATE INDUCED | ONSET OF CLINICAL MANIFESTATIONS | PERSISTENCE OF DAI CLINICAL MANIFESTATIONS | |
| FOCAL INJURY | DIFFUSE AXONAL INJURY (DAI) | |||
| Mild blunt trauma | Mild concussion | Immediate | Hours to days | |
| Moderate blunt trauma | Classic cerebral concussion | Immediate | Up to 6 months or longer | |
| Paraplegia (associated with injury to top of head) | Immediate | |||
| Blindness (associated with occipital injury) | Immediate | |||
| Delayed development of unresponsiveness (vasomotor or vasovagal syncopal episode) | Delayed | |||
| Severe blunt trauma | Mild DAI | Immediate | Recovery in days to weeks | |
| Moderate DAI | Immediate | Residual manifestation | ||
| Severe DAI | Immediate | Permanent severe disability | ||
| Acute epidural hemorrhage | Immediate to delayed (2-3 hours) | |||
| Acute contusional swelling | Delayed onset (few hours after injury) | |||
| Acute subdural hematoma | Delayed onset (few hours to 1 week after injury) | |||
| Subacute subdural hematoma∗ | Delayed onset (1 to few weeks) | |||
| Subdural hygroma (fluid accumulation) | Delayed onset | |||
| Traumatic cerebral hemorrhage∗ | Delayed onset (as late as 1 week after injury) | |||
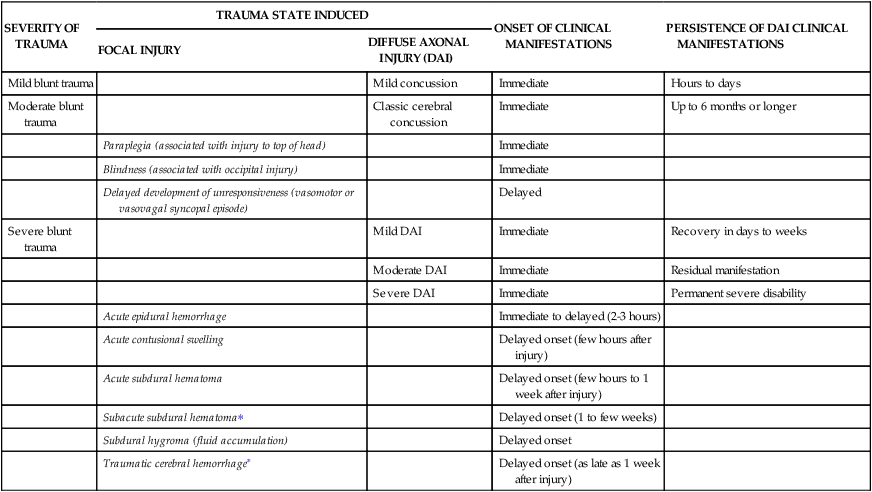
∗May be seen after moderate head injury, especially in elderly people.
The Glasgow Coma Scale (GCS) is used to assess severity of injury.4 The hallmark of a severe TBI is loss of consciousness for 6 hours or more. TBI classifications using the GCS are: (1) mild TBI with GCS score of 13 to 15, associated with mild concussion; (2) moderate TBI with GCS score of 9 to 12, associated with structural injury such as hemorrhage or contusion; and (3) severe TBI with GCS score of 3 to 8, associated with cognitive and/or physical disability or death. Age and admission GCS score are important diagnostic factors in TBI.5 Mild TBI is characterized by five features: a GCS score between 13 and 15, a brief period of unconsciousness or dazed consciousness, posttraumatic amnesia, negative neuroimaging studies, and recovery within weeks to months without sequelae or the need for specific medical intervention.
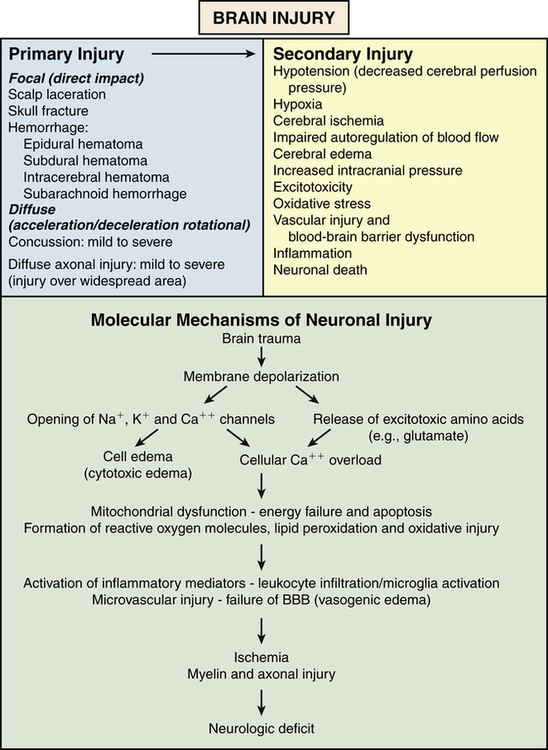
Three mechanisms produce brain damage: primary, secondary, and tertiary injury. Primary injury is caused by the impact and involves neural injury, primary glial injury, vascular responses, and shearing and rotational forces. Secondary injury is an indirect consequence of the primary injury and includes a cascade of cellular and molecular brain events (Figure 18-1). Tertiary injury can develop days or months later as a consequence of systemic complications, such as pneumonia, fever, infections, and immobility, which contribute to further brain injury or delays in repair.
Primary Brain Injury
Primary brain injuries can be classified as focal, affecting one area of the brain, or diffuse (diffuse axonal injury [DAI]), involving more than one area of the brain (see Table 18-1). Both types of injury can be associated with the same initiating event and can occur with brain trauma or stroke syndromes (see Figure 18-1). Focal brain injury accounts for more than two thirds of head injury deaths; DAI accounts for less than one third. However, more severely disabled survivors, including those in an unresponsive state or reduced level of consciousness, have DAI.
Focal Brain Injury
Focal brain injury results from compression of the skull at the point of impact and rebound effects. The focal injury may be a coup injury (directly below the point of impact) or contrecoup (on the pole opposite the site of impact) (Figure 18-2). Objects (e.g., baseball bat, weapon) striking the front of the head usually produce only coup injuries (contusions and fractures) because the inner skull in the occipital area is smooth. Objects striking the back of the head usually result in both coup and contrecoup injuries because of the irregularity of the inner surface of the frontal bones. Objects striking the side of the head may produce coup or contrecoup injuries. The force of impact (translational acceleration) typically produces localized brain contusions. Injury to the vault, other vessels, and supporting structures can produce epidural hemorrhage and subdural and intracerebral hematomas.
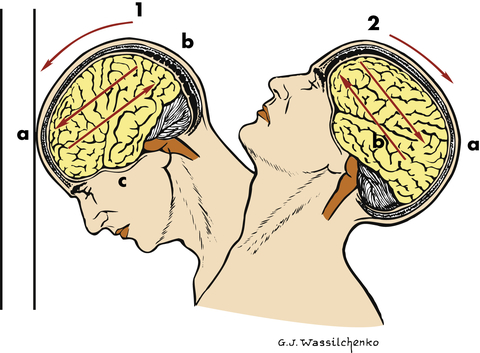
1, Coup injury: impact against object; a, site of impact and direct trauma to brain; b, shearing of subdural veins; c, trauma to base of brain. 2, Contrecoup injury: impact within skull; a, site of impact from brain hitting opposite side of skull; b, shearing forces through brain. These injuries occur in one continuous motion—the head strikes the wall (coup) and then rebounds (contrecoup). (Modified from Rudy EB: Advanced neurological and neurosurgical nursing, St Louis, 1984, Mosby.)
Contusions (blood leaking from an injured vessel) (Figure 18-3) are found most commonly in the frontal lobes, particularly at the poles and along the inferior orbital surfaces; in the temporal lobes, especially at the anterior poles and along the inferior surface; and at the frontotemporal junction. Contusion severity varies with the amount of impact energy transmitted by the skull to underlying brain tissue. The smaller the area of impact, the greater the severity of injury because the force is concentrated into a smaller area. Brain edema forms around and in damaged neural tissues, contributing to increasing intracranial pressure (ICP). Within the contused areas are infarction and necrosis, multiple hemorrhages, and edema. The tissue has a pulpy quality. The maximum effects of injury related to contusion, bleeding, and edema peak 18 to 36 hours after severe head injury.
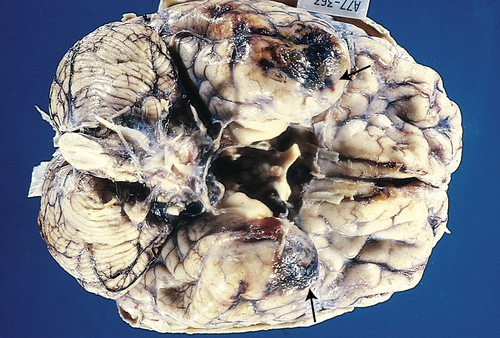
The temporal poles are discolored by areas of hemorrhage (arrows). Such lesions represent “bruises” on the surface of the brain caused by violent contact between the delicate brain parenchyma and the hard inner surface of the skull. (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders.)
Evaluation is based on history, level of consciousness using the Glasgow Coma Scale, imaging studies (e.g., computed tomography [CT], magnetic resonance imaging [MRI], and positron-emission tomography [PET]), and assessment of vital parameters (e.g., ICP and EEG). Large contusions and lacerations with hemorrhage may be excised surgically.6 Otherwise, treatment is directed at controlling ICP and managing symptoms.
An artery is the source of bleeding in 85% of extradural hematomas (Figure 18-4); 15% result from injury to the meningeal vein or dural sinus. Ninety percent of individuals also have a skull fracture. The temporal fossa is the most common site of extradural hematoma caused by injury to the middle meningeal artery or vein. The resulting shift of the temporal lobe medially precipitates uncal and hippocampal gyrus herniation through the tentorial notch. Extradural hemorrhages are found occasionally in the subfrontal area (especially in the young and older adult populations), caused by injury to the anterior meningeal artery or a venous sinus, and in the occipital-suboccipital area, which results in herniation of the posterior fossa contents through the foramen magnum. CT and MRI show a lens-shaped mass over the surface of the cortex.
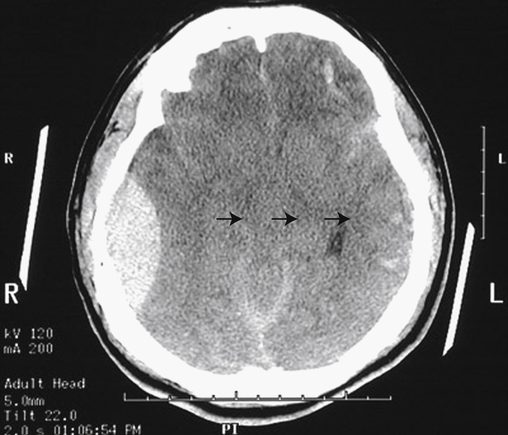
Note the large right epidural hematoma with a lens-shaped outline as the smooth dura becomes indented against the underlying cortex on the right lateral aspect of the cerebrum. The epidural hematoma is confined within an area bounded by cranial sutures where the dura is firmly adherent to the skull. Note the mass effect with effacement of the lateral ventricles and the shift of midline to the left (arrows). In this case the individual fell from a height and struck the right side of his head, severing the middle meningeal artery. This epidural hematoma collected within hours. CT, Computed tomography. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
Subdural hematomas arise in 10% to 20% of TBIs. MVAs are the most common cause of subdural hematomas; 50% of subdural hematomas are associated with skull fractures. Falls, especially in older adults or in those with long-term alcohol abuse, are associated with chronic subdural hematomas.
Acute subdural hematomas rapidly develop (within 48 hours) and usually are located at the top of the skull (the cerebral convexities). On CT they appear as a high-density mass. Bilateral hematomas occur in 15% to 20% of persons. Subacute subdural hematomas develop more slowly, often over 48 hours to 2 weeks. On CT they appear as a mixed-density mass. Chronic subdural hematomas (commonly found in older adults and those who abuse alcohol and have some degree of brain atrophy with a subsequent increase in the extradural space) develop over weeks to months. Tearing of the bridging veins is the major cause of rapidly developing and subacutely developing subdural hematomas, although torn cortical veins or venous sinuses and contused tissue may be the source. These subdural hematomas act as expanding masses, giving rise to increased ICP that eventually compresses the bleeding vessels (Figures 18-5 and 18-6). The displacement of brain tissue can result in a herniation syndrome.
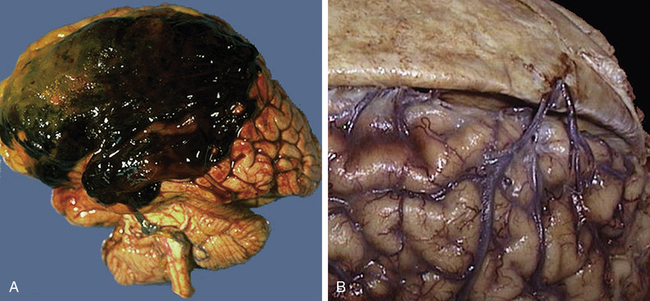
A large subdural hematoma (A) is seen in the frontoparietal region. A subdural hematoma forms after head trauma that severs the bridging veins from dura to brain, shown in the right panel (B), where the dura has been reflected to reveal the normal appearance of the bridging veins that extend across to the superior aspect of the cerebral hemispheres. Older adults and the very young are at greater risk because their cerebral veins are more vulnerable to injury. Because the bleeding is venous, blood collects over hours to weeks, with variable onset of symptoms. Because the blood collects beneath the dura, a subdural hematoma can be seen to cross the region of cranial sutures. (From Klatt EC: Robbins and Cotran atlas of pathology, Philadelphia, 2006, Saunders.)
Compression of underlying brain and lateral ventricle. (From Kissane JM, editor: Anderson’s pathology, ed 9, St Louis, 1993, Mosby.)
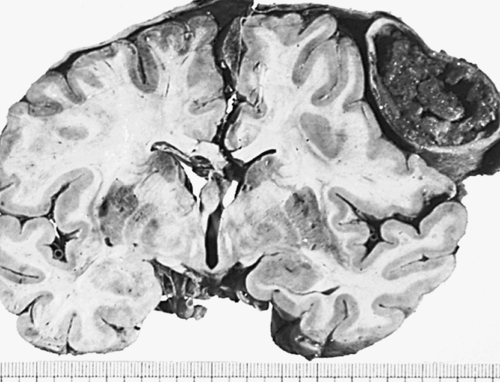
Intracerebral hematomas occur in 2% to 3% of head injuries and are usually associated with MVAs and falls from some distance. Intracerebral hematomas may be single or multiple, and they are associated with contusions. Although most commonly located in the frontal and temporal lobes, intracerebral hematomas may occur in the hemispheric deep white matter. Small blood vessels are traumatized by penetrating injury or shearing forces. The intracerebral hematoma then acts as an expanding mass, resulting in increased ICP and compression of brain tissues with resultant edema and ischemia (Figure 18-7). Delayed intracerebral hematomas may appear 3 to 10 days after the head injury.
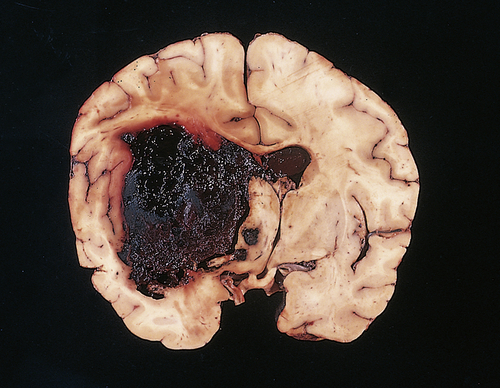
A fresh hematoma has disrupted and expanded the left cerebral hemisphere, causing the midline structures to shift to the right. Uncontrolled hypertension is an important cause of this catastrophic lesion. (From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders.)
Evacuation of a singular intracerebral hematoma has only occasionally been helpful, mostly for subcortical white matter hematomas. Otherwise, treatment is directed at reducing the ICP to maintain cerebral perfusion pressure and allowing the hematoma to reabsorb slowly. Secondary brain injury is common (see p. 588).
Stretch injury involves blood vessels and nerves that are damaged without direct contact as a result of the amount of tissue stretched secondary to shape, deformation, and striking velocity. Air compressed in front of a bullet exerts an explosive effect on entry, producing extreme distant tissue damage and an immediate primary increase in ICP; a cavity many times greater than the size of the bullet is produced because the brain tissue is propelled away from the tract. The cavity and pressure produce contrecoup injuries. The intracranial volume is increased directly by the projectile and the debris. The temporary cavity collapses back onto itself, leaving a smaller, permanent cavity. Intracranial bleeding occurs into the permanent cavity and may cause the cavity to expand. Edema in and around the injured brain tissue rapidly develops; edema and bleeding contribute markedly to increased ICP. This second rise in ICP to 60 to 100 mmHg may last 2 to 5 minutes. Because of acute ischemic damage to the tract, necrosis of tissue begins. Within hours after bullet-induced injury, tissue within 1 cm adjacent to the tract disintegrates. Demyelination of white matter affected by hemorrhage and edema occurs by the second day. Clinical manifestations such as unconsciousness, flaccidity, or decerebrate posture (see Chapter 17) are associated with high mortality.
A compound linear fracture is débrided nonsurgically in cooperative adults and surgically in children and uncooperative adults. Cranioplasty with insertion of bone or an artificial graft may be necessary but often is delayed until antibiotics have been given. Antibiotics are administered after surgery. Open-head injury often requires surgery to débride the traumatized tissues to prevent infection and to remove blood clots to help reduce the ICP. ICP also is managed with dehydrating agents, osmotic diuretics, or a combination of these drugs. Broad-spectrum antibiotics are administered to prevent infection. Use of prophylactic antibiotics is controversial because studies have failed to demonstrate that they reduce the rate of infection.7 Bed rest and close observation for meningitis and other complications are prescribed for a basilar skull fracture.
Diffuse Brain Injury
Diffuse brain injury (diffuse axonal injury [DAI]) involves widespread areas of the brain. Damage to delicate axonal fibers and white matter tracts that project to the cerebral cortex causes concussion. Mechanical effects from high levels of acceleration and deceleration (whiplash) and rotational and twisting movements are the primary mechanisms of injury, producing strains and distortions within the brain (see Figure 18-2). As a result of the skull’s motion caused by its attachment to the neck, striking the head causes rotational forces that can trigger shearing forces on brain tissues. Stretching and tearing of nerve fibers result in axonal damage. The most severe axonal injuries are located more peripheral to the brainstem, causing extensive cognitive and affective impairments, as seen in survivors of traumatic brain injury from vehicular crashes. Axonal damage reduces the speed of information processing and response and disrupts the individual’s attention span.
Pathophysiologically, axonal damage can be seen only with an electron microscope and involves numerous axons alone or axonal injury in conjunction with actual tissue tears (Figure 18-8). Areas where axons and small blood vessels are torn appear as small hemorrhages, located particularly in the corpus callosum and dorsolateral quadrant of the rostral brainstem at the superior cerebellar peduncle. Increasingly more damaged axons are visible 12 hours to several days after the injury and secondary brain injury also is occurring (see p. 588). The severity of diffuse injury correlates with how much shearing force was applied to the brain. DAI is not associated with intracranial hypertension immediately after injury; however, secondary brain injury causes acute brain swelling from increased intravascular blood flow within the brain, vasodilation, and increased cerebral blood volume.
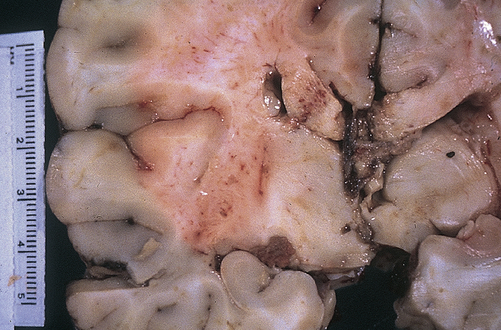
Gross photograph demonstrating characteristic hemorrhage lesions within the corpus callosum. (Courtesy Walter Kemp, MD, Department of Pathology, University of Texas Southwestern Medical School, Dallas. From Kumar V, Cotran RS, Robbins SL: Robbins basic pathology, ed 7, Philadelphia, 2003, Saunders.)
Several categories of diffuse brain injury exist: mild concussion, classic concussion, mild DAI, moderate DAI, and severe DAI (see Table 18-1 and Figure 18-1).
Mild concussion (mild traumatic brain injury) is characterized by immediate but transitory clinical manifestations. CSF pressure rises, and ECG and EEG changes occur without loss of consciousness. The Glasgow Coma Scale score is 13 to 15.8 The initial confusional state lasts from 1 to several minutes, possibly with amnesia for events preceding the trauma (retrograde amnesia). Anterograde amnesia also may exist transiently. Persons may experience head pain and complain of nervousness and “not being themselves” for up to a few days. Three grades have been described:
Grade I: Transient confusion and disorientation accompanied by amnesia (momentary); no loss of consciousness; symptoms resolve within 15 minutes
Grade II: Transient confusion and retrograde amnesia that develops after 5 to 10 minutes (memory loss involves only events occurring several minutes before injury); symptoms last more than 15 minutes
Grade III: Any loss of consciousness (seconds or minutes); confusion and retrograde and anterograde amnesia remain present from impact and persist for several minutes
Some of the effects of a concussion may persist for weeks or months, depending on the severity of the injury. Approximately 15% to 30% of persons have a postconcussive syndrome that includes headache, nervousness or anxiety, irritability, insomnia, depression, inability to concentrate, forgetfulness, and fatigability.9 Treatment entails reassurance, symptomatic relief, and close observation for 24 hours with follow-up care for persistent symptoms.
DAI produces prolonged traumatic coma lasting more than 6 hours because of axonal disruption. Three forms of DAI exist: mild, moderate, and severe. In mild diffuse axonal injury, posttraumatic coma lasts 6 to 24 hours. Death is uncommon but residual cognitive, psychologic, and sensorimotor deficits may persist. Mild DAI is a relatively uncommon lesion, occurring in 8% of all severe head injuries and 19% of all cases of DAI. In mild DAI 30% of persons display decerebrate or decorticate posturing (see Figure 17-6); they may experience prolonged periods of stupor or restlessness.
Severe diffuse axonal injury involves severe mechanical disruption of many axons in both cerebral hemispheres and those extending to the diencephalon and brainstem. Severe DAI represents 16% of all severe head injuries and 36% of all cases of DAI. With an initial GCS score of 3, the mortality is 78%;5 with an initial score between 3 and 8, the mortality is 36%; 16% of persons have either a moderate or a severe disability and 5% survive in a coma (unresponsive) state.5
High-resolution CT scans and MRIs assist in the diagnosis of focal and diffuse brain injuries.5,10 The goal of treatment is to maintain cerebral perfusion and oxygenation and promote neuroprotection. Implementation of traumatic brain injury guidelines decreases deaths and improves neurologic outcome.11,12 Specifically, the evidence is inconclusive about the effectiveness of hyperventilation and mild hypothermia. Hypertonic saline or mannitol is used selectively to manage increased intracranial pressure (IICP).13 Barbiturates have not been shown to be effective in reducing intracranial pressure or preventing adverse outcomes after TBI. The Corticosteroid Randomisation After Significant Head Injury (CRASH) trial showed corticosteroids increase mortality with acute TBI, so these drugs are no longer used. Antiepileptic drugs may be recommended to reduce the onset of early and late seizures. Prophylactic antibiotics have not been shown to reduce the risk of meningitis or death with a skull fracture. Progesterone is under investigation and may improve neurologic outcome.14 Extensive research is underway to discover effective therapeutic interventions. Fluid and nutrition management has emerged as critically important in the care of individuals with severe brain injuries.15
Secondary Brain Injury
Secondary neuronal injury occurs when there is brain or spinal cord trauma and in stroke syndromes. It occurs within hours to days of the primary injury. Secondary brain injury is the result of both systemic and intracranial processes and is an indirect result of primary brain injury. Systemic hypotension, hypoxia, anemia, hypoglycemia, hyperglycemia, and hypercapnia contribute to secondary brain insults. Mechanisms of secondary injury include altered cerebral blood flow, ischemia, inflammation, cerebral edema, decreased cerebral perfusion pressure (CPP), IICP, and brain herniation. Molecular alterations occur with stretching and tearing of axonal membranes and ischemia (see Figure 18-1). The complex pathophysiology of secondary brain injury underlies the functional, cognitive, and behavioral changes associated with brain injury and the motor and sensory losses associated with spinal cord injury.
Widespread release of excitatory neurotransmitters (e.g., glutamate and aspartate) causes calcium and sodium influx that is neurotoxic (excitotoxicity). Mitochondrial loading with calcium leads to mitochondrial failure, anaerobic metabolism, lactic acid production, and increased levels of reactive oxygen species (ROS), phospholipases, and other enzymes. ROS and phospholipase damage proteins and the phospholipid components of cell and organelle membranes, causing cell swelling, vacuolization, and, ultimately, necrotic or programmed cell death (apoptosis). Energy metabolism is depressed with alteration of gene expression. These events lead to failure of the capillary blood-brain barrier and vasogenic edema. The influx of sodium attracts water and results in intracellular cytotoxic edema in neurons, astrocytes, and microglia irrespective of the integrity of the vascular endothelial wall. Edema increases intracranial pressure, contributing to tissue hypoxia and cerebral ischemia.16,17
Both primary and secondary injuries cause inflammation (see Chapter 7), which occurs immediately and persists for weeks. Inflammation is critical for wound healing; immune cells, including neutrophils, macrophages, and T-cell lymphocytes, need to infiltrate injured tissue and clear debris, thus promoting formation of scar tissue. However, an overactive response leads to occlusion of the microvasculature with leukocytes and platelets. The release of inflammatory cytokines (e.g., tumor necrosis factor-alpha [TNF-α], interleukins, and interferons), proteases, free radicals, prostaglandins, and complement alters the blood-brain barrier and causes vasoconstriction, brain edema, increased intracranial pressure, reduced cerebral perfusion, and ischemia, aggravating secondary brain injury.17,18
Intracranial hemorrhage, whether traumatic or related to a stroke syndrome, may contribute to secondary brain injury. The bleeding is often accompanied by increased ICP, ischemia, oxidative damage, and vasogenic and cytotoxic edema as previously described. In addition, the vascular injury can cause activation of platelets, which contribute to vasoconstriction in large arteries and vasodilation in small arteries. Activated platelets also trigger the release of free radicals from granulocytes, enhancing oxidative stress. Vascular injury causes coagulation disorders from release of increased amounts of tissue factor from endothelial cells, which initiates the coagulation cascade and overwhelms the normal factors that control coagulation. Free heme and free iron are released from red blood cells and both cause the induction of free radicals, lipid peroxidation, and nerve damage.19
The management of secondary brain trauma is related to prevention and includes removal of hematomas and management of hypotension, hypoxemia, anemia, intracranial pressure, body temperature, ventilation, fluid replacement, and nutrition. Research is in progress to find specific pharmacologic interventions and neuroprotective agents that limit the progression of secondary injury and to identify biomarkers to guide prevention, prognosis, and treatment20,21 (see What’s New? Progesterone for Treatment of Traumatic Brain Injury [TBI]).
Genetics of Head Injury
Certain genes are up-regulated and others are down-regulated after head trauma. The attention of researchers has been focused predominantly on the apolipoprotein E (apoE) gene and its various alleles. Certain alleles have been correlated with increased susceptibility to and severity of head injury. Other alleles have been associated with improved or diminished recovery after head injury. The clinical significance of these findings is not clearly known.22,23
Spinal Cord Trauma
The number of spinal cord injuries (SCIs) in the United States is approximately 273,000, with 12,000 new cases annually; of these, 80.7% are males, mostly young adults. The average age is 40.7 years. Since 2010 the causes include vehicular (36.5%), falls (28%), violence (14%), and sports (9%). The extent of injury has been 41% incomplete quadriplegia, 16% complete quadriplegia, 18.7% incomplete paraplegia, and 18% complete paraplegia.24
Pathophysiology
Primary spinal cord injury occurs with the initial mechanical trauma and immediate tissue destruction. Vertebral fractures or dislocations can cause bone fragments or connective tissues to compress and damage nerve fibers. Vertebral injuries result from acceleration, deceleration, or deformation forces at impact. These forces cause compression, traction, or shearing of tissues. The bones, ligaments, joints, and neural tissue of the vertebral column may be damaged by hyperextension, hyperflexion, vertical compression, or rotation (Figures 18-9 through 18-12). Vertebral fracture and often compression of one or more elements, dislocation of its elements, or both fracture and dislocation can occur (see Figure 15-17 for the structure of the vertebral column). Vertebral injuries can be classified as (1) simple fracture, a single break usually affecting transverse or spinous processes; (2) compressed (wedged) vertebral fracture, in which a vertebral body is compressed anteriorly; (3) comminuted (burst) fracture, in which a vertebral body is shattered into several fragments; and (4) dislocation.
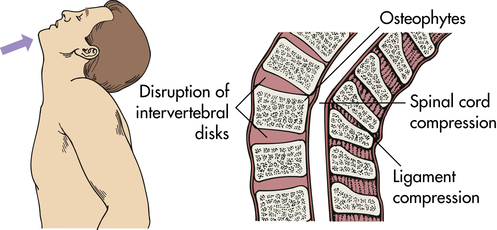
Hyperextension can result in fracture or nonfracture injuries with spinal cord damage.
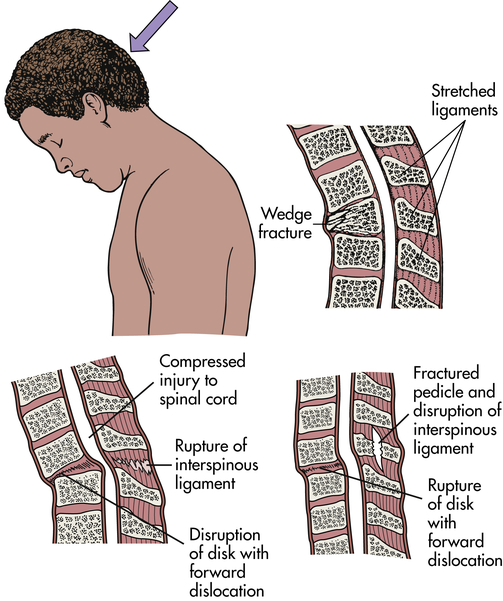
Hyperflexion produces translation (subluxation) of vertebrae, which compromises the central canal and compresses spinal cord parenchyma or vascular structures.
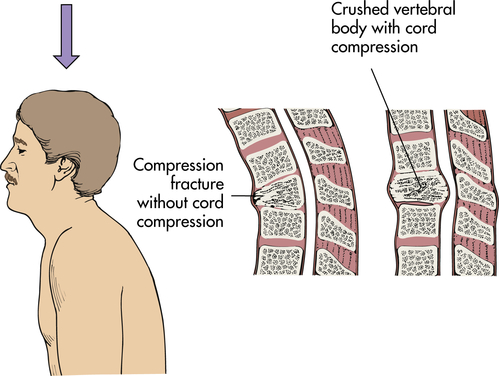
In axial compression the spinal cord is contused directly by retropulsion of bone or disk material into the spinal canal.
Vertebrae fracture readily with direct and indirect trauma. When the supporting ligaments are torn, the vertebrae move out of alignment, and dislocations occur. A horizontal force moves the vertebrae straight forward; if the individual is in a flexed position at the time of injury, the vertebrae are then in an angulated position. Flexion and extension injuries may result in dislocations. (Mechanisms of vertebral injury are presented in Table 18-2.) Vertebral injuries occur mostly at vertebrae C1-C2 (cervical), C4-C7, and T1-L2 (thoracic-lumbar) (see Figure 15-17). These are the most mobile portions of the vertebral column. The cord occupies most of the vertebral canal in the cervical and lumbar regions. The size makes the cord in these areas more easily injured. The primary spinal cord injuries are summarized in Table 18-3. Noncontiguous vertebral injuries are common. Further, primary injury occurs if an injured spine is not adequately immobilized. Primary spinal cord injury may occur in the absence of vertebral fracture or dislocation from longitudinal stretching of the cord with or without flexion and/or extension of the vertebral column. The stretching causes altered axon transport, edema, myelin degeneration, and retrograde or wallerian degeneration.
TABLE 18-2
MECHANISMS OF VERTEBRAL INJURY
| MECHANISMS OF INJURY | VERTEBRAL INJURY | FORCES OF INJURY | LOCATION OF INJURY |
| Hyperextension | Fracture and dislocation of posterior elements such as spinous processes, transverse processes, laminae, pedicles, or posterior ligaments | Results from forces of acceleration-deceleration and the sudden reduction in the anteroposterior diameter of the spinal cord | Cervical area |
| Hyperflexion | Fracture or dislocation of the vertebral bodies, disks, or ligaments | Results from sudden and excessive force that propels the neck forward or causes an exaggerated lateral movement of the neck to one side | Cervical area |
| Vertical compression (axonal loading) | Shattering fractures | Results from a force applied along an axis from the top of the cranium through the vertebral bodies | T12-L2 |
| Rotational forces (flexion-rotation) | Ruptures support ligaments in addition to producing fractures | Adds shearing force to acceleration-deceleration forces | Cervical area |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


