140 | Disorders of Platelets and Vessel Wall |
Hemostasis is a dynamic process in which the platelet and the blood vessel wall play key roles. Platelets become activated upon adhesion to von Willebrand factor (VWF) and collagen in the exposed subendothelium after injury. Platelet activation is also mediated through shear forces imposed by blood flow itself, particularly in areas where the vessel wall is diseased, and is also affected by the inflammatory state of the endothelium. The activated platelet surface provides the major physiologic site for coagulation factor activation, which results in further platelet activation and fibrin formation. Genetic and acquired influences on the platelet and vessel wall, as well as on the coagulation and fibrinolytic systems, determine whether normal hemostasis or bleeding or clotting symptoms will result.
THE PLATELET
Platelets are released from the megakaryocyte, likely under the influence of flow in the capillary sinuses. The normal blood platelet count is 150,000–450,000/μL. The major regulator of platelet production is the hormone thrombopoietin (TPO), which is synthesized in the liver. Synthesis is increased with inflammation and specifically by interleukin 6. TPO binds to its receptor on platelets and megakaryocytes, by which it is removed from the circulation. Thus a reduction in platelet and megakaryocyte mass increases the level of TPO, which then stimulates platelet production. Platelets circulate with an average life span of 7–10 days. Approximately one-third of the platelets reside in the spleen, and this number increases in proportion to splenic size, although the platelet count rarely decreases to <40,000/μL as the spleen enlarges. Platelets are physiologically very active, but are anucleate, and thus have limited capacity to synthesize new proteins.
Normal vascular endothelium contributes to preventing thrombosis by inhibiting platelet function (Chap. 78). When vascular endothelium is injured, these inhibitory effects are overcome, and platelets adhere to the exposed intimal surface primarily through VWF, a large multimeric protein present in both plasma and in the extracellular matrix of the subendothelial vessel wall. Platelet adhesion results in the generation of intracellular signals that lead to activation of the platelet glycoprotein (Gp) IIb/IIIa (αIIbβ3) receptor and resultant platelet aggregation.
Activated platelets undergo release of their granule contents, which include nucleotides, adhesive proteins, growth factors, and procoagulants that serve to promote platelet aggregation and blood clot formation and influence the environment of the forming clot. During platelet aggregation, additional platelets are recruited to the site of injury, leading to the formation of an occlusive platelet thrombus. The platelet plug is stabilized by the fibrin mesh that develops simultaneously as the product of the coagulation cascade.
THE VESSEL WALL
Endothelial cells line the surface of the entire circulatory tree, totaling 1–6 × 1013 cells, enough to cover a surface area equivalent to about six tennis courts. The endothelium is physiologically active, controlling vascular permeability, flow of biologically active molecules and nutrients, blood cell interactions with the vessel wall, the inflammatory response, and angiogenesis.
The endothelium normally presents an antithrombotic surface (Chap. 78) but rapidly becomes prothrombotic when stimulated, which promotes coagulation, inhibits fibrinolysis, and activates platelets. In many cases, endothelium-derived vasodilators are also platelet inhibitors (e.g., nitric oxide) and, conversely, endothelium-derived vasoconstrictors (e.g., endothelin) can also be platelet activators. The net effect of vasodilation and inhibition of platelet function is to promote blood fluidity, whereas the net effect of vasoconstriction and platelet activation is to promote thrombosis. Thus, blood fluidity and hemostasis are regulated by the balance of antithrombotic/prothrombotic and vasodilatory/vasoconstrictor properties of endothelial cells.
DISORDERS OF PLATELETS
THROMBOCYTOPENIA
Thrombocytopenia results from one or more of three processes: (1) decreased bone marrow production; (2) sequestration, usually in an enlarged spleen; and/or (3) increased platelet destruction. Disorders of production may be either inherited or acquired. In evaluating a patient with thrombocytopenia, a key step is to review the peripheral blood smear and to first rule out “pseudothrombocytopenia,” particularly in a patient without an apparent cause for the thrombocytopenia. Pseudothrombocytopenia (Fig. 140-1B) is an in vitro artifact resulting from platelet agglutination via antibodies (usually IgG, but also IgM and IgA) when the calcium content is decreased by blood collection in ethylenediamine tetraacetic (EDTA) (the anticoagulant present in tubes [purple top] used to collect blood for complete blood counts [CBCs]). If a low platelet count is obtained in EDTA-anticoagulated blood, a blood smear should be evaluated and a platelet count determined in blood collected into sodium citrate (blue top tube) or heparin (green top tube), or a smear of freshly obtained unanticoagulated blood, such as from a finger stick, can be examined.
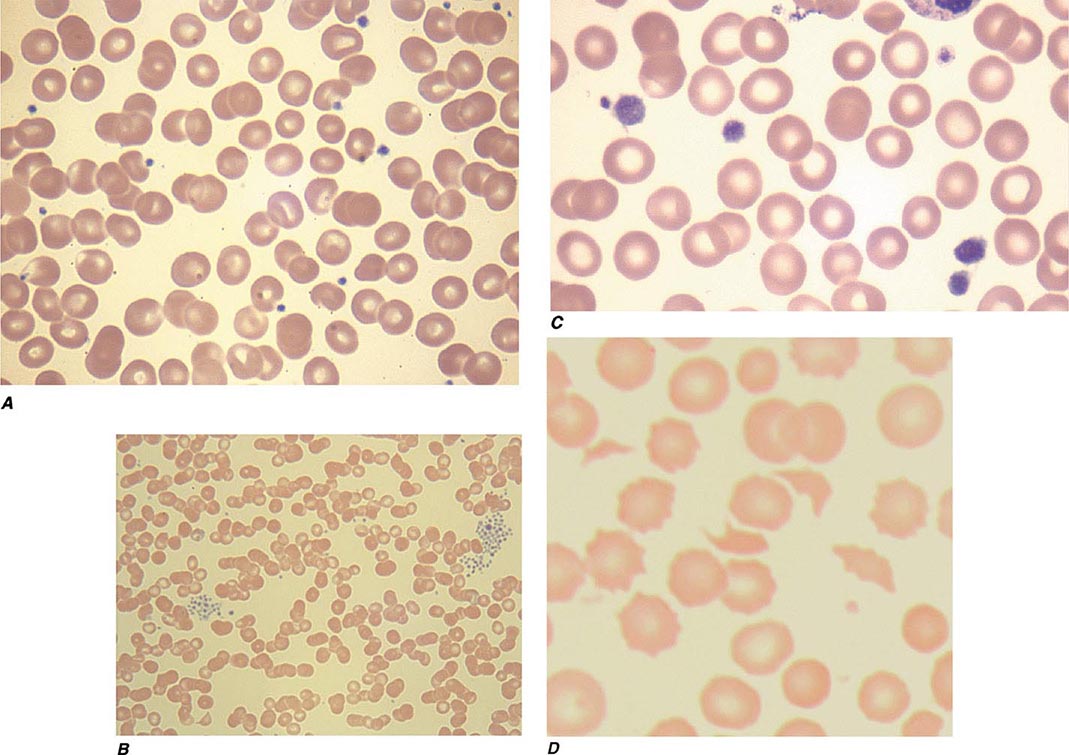
FIGURE 140-1 Photomicrographs of peripheral blood smears. A. Normal peripheral blood. B. Platelet clumping in pseudothrombocytopenia. C. Abnormal large platelet in autosomal dominant macrothrombocytopenia. D. Schistocytes and decreased platelets in microangiopathic hemolytic anemia.
APPROACH TO THE PATIENT:
Thrombocytopenia
The history and physical examination, results of the CBC, and review of the peripheral blood smear are all critical components in the initial evaluation of thrombocytopenic patients (Fig. 140-2). The overall health of the patient and whether he or she is receiving drug treatment will influence the differential diagnosis. A healthy young adult with thrombocytopenia will have a much more limited differential diagnosis than an ill hospitalized patient who is receiving multiple medications. Except in unusual inherited disorders, decreased platelet production usually results from bone marrow disorders that also affect red blood cell (RBC) and/or white blood cell (WBC) production. Because myelodysplasia can present with isolated thrombocytopenia, the bone marrow should be examined in patients presenting with isolated thrombocytopenia who are older than 60 years of age. While inherited thrombocytopenia is rare, any prior platelet counts should be retrieved and a family history regarding thrombocytopenia obtained. A careful history of drug ingestion should be obtained, including nonprescription and herbal remedies, because drugs are the most common cause of thrombocytopenia.
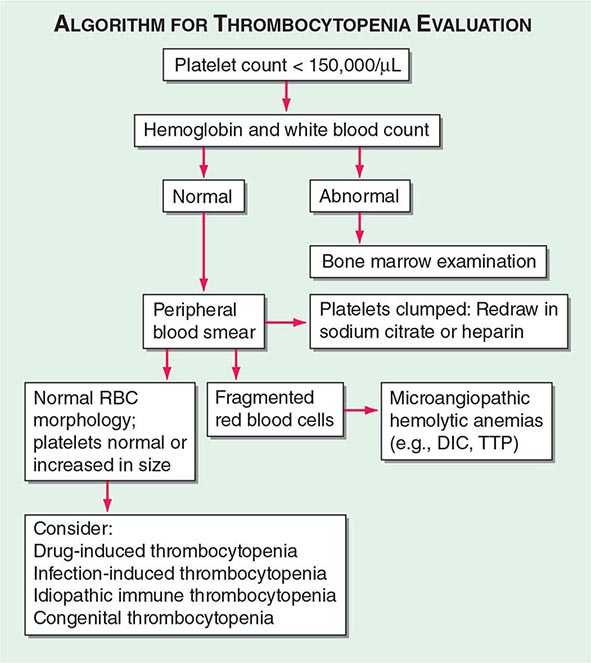
FIGURE 140-2 Algorithm for evaluating the thrombocytopenic patient. DIC, disseminated intravascular coagulation; RBC, red blood cell; TTP, thrombotic thrombocytopenic purpura.
The physical examination can document an enlarged spleen, evidence of chronic liver disease, and other underlying disorders. Mild to moderate splenomegaly may be difficult to appreciate in many individuals due to body habitus and/or obesity but can be easily assessed by abdominal ultrasound. A platelet count of approximately 5000–10,000 is required to maintain vascular integrity in the microcirculation. When the count is markedly decreased, petechiae first appear in areas of increased venous pressure, the ankles and feet in an ambulatory patient. Petechiae are pinpoint, nonblanching hemorrhages and are usually a sign of a decreased platelet number and not platelet dysfunction. Wet purpura, blood blisters that form on the oral mucosa, are thought to denote an increased risk of life-threatening hemorrhage in the thrombocytopenic patient. Excessive bruising is seen in disorders of both platelet number and function.
Infection-Induced Thrombocytopenia Many viral and bacterial infections result in thrombocytopenia and are the most common noniatrogenic cause of thrombocytopenia. This may or may not be associated with laboratory evidence of disseminated intravascular coagulation (DIC), which is most commonly seen in patients with systemic infections with gram-negative bacteria. Infections can affect both platelet production and platelet survival. In addition, immune mechanisms can be at work, as in infectious mononucleosis and early HIV infection. Late in HIV infection, pancytopenia and decreased and dysplastic platelet production are more common. Immune-mediated thrombocytopenia in children usually follows a viral infection and almost always resolves spontaneously. This association of infection with immune thrombocytopenic purpura is less clear in adults.
Bone marrow examination is often requested for evaluation of occult infections. A study evaluating the role of bone marrow examination in fever of unknown origin in HIV-infected patients found that for 86% of patients, the same diagnosis was established by less invasive techniques, notably blood culture. In some instances, however, the diagnosis can be made earlier; thus, a bone marrow examination and culture are recommended when the diagnosis is needed urgently or when other, less invasive methods have been unsuccessful.
Drug-Induced Thrombocytopenia Many drugs have been associated with thrombocytopenia. A predictable decrease in platelet count occurs after treatment with many chemotherapeutic drugs due to bone marrow suppression (Chap. 103e). Drugs that cause isolated thrombocytopenia and have been confirmed with positive laboratory testing are listed in Table 140-1, but all drugs should be suspect in a patient with thrombocytopenia without an apparent cause and should be stopped, or substituted, if possible. A helpful website, Platelets on the Internet (http://www.ouhsc.edu/platelets/ditp.html), lists drugs and supplements reported to have caused thrombocytopenia and the level of evidence supporting the association. Although not as well studied, herbal and over-the-counter preparations may also result in thrombocytopenia and should be discontinued in patients who are thrombocytopenic.
DRUGS REPORTED AS DEFINITELY OR PROBABLY CAUSING ISOLATED THROMBOCYTOPENIAa |
aBased on scoring requiring a compatible clinical picture and positive laboratory testing.
Source: Adapted from DM Arnold et al: J Thromb Hemost 11:169, 2013.
Classic drug-dependent antibodies are antibodies that react with specific platelet surface antigens and result in thrombocytopenia only when the drug is present. Many drugs are capable of inducing these antibodies, but for some reason, they are more common with quinine and sulfonamides. Drug-dependent antibody binding can be demonstrated by laboratory assays, showing antibody binding in the presence of, but not without, the drug present in the assay. The thrombocytopenia typically occurs after a period of initial exposure (median length 21 days), or upon reexposure, and usually resolves in 7–10 days after drug withdrawal. The thrombocytopenia caused by the platelet Gp IIb/IIIa inhibitory drugs, such as abciximab, differs in that it may occur within 24 h of initial exposure. This appears to be due to the presence of naturally occurring antibodies that cross-react with the drug bound to the platelet.
Heparin-Induced Thrombocytopenia Drug-induced thrombocytopenia due to heparin differs from that seen with other drugs in two major ways. (1) The thrombocytopenia is not usually severe, with nadir counts rarely <20,000/μL. (2) Heparin-induced thrombocytopenia (HIT) is not associated with bleeding and, in fact, markedly increases the risk of thrombosis. HIT results from antibody formation to a complex of the platelet-specific protein platelet factor 4 (PF4) and heparin. The anti-heparin/PF4 antibody can activate platelets through the FcγRIIa receptor and also activate monocytes and endothelial cells. Many patients exposed to heparin develop antibodies to heparin/PF4, but do not appear to have adverse consequences. A fraction of those who develop antibodies will develop HIT, and a portion of those (up to 50%) will develop thrombosis (HITT).
HIT can occur after exposure to low-molecular-weight heparin (LMWH) as well as unfractionated heparin (UFH), although it is more common with the latter. Most patients develop HIT after exposure to heparin for 5–14 days (Fig. 140-3). It occurs before 5 days in those who were exposed to heparin in the prior few weeks or months (<~100 days) and have circulating anti-heparin/PF4 antibodies. Rarely, thrombocytopenia and thrombosis begin several days after all heparin has been stopped (termed delayed-onset HIT). The “4T’s” have been recommended to be used in a diagnostic algorithm for HIT: thrombocytopenia, timing of platelet count drop, thrombosis and other sequelae such as localized skin reactions, and other causes of thrombocytopenia not evident. Application of the 4T scoring system is very useful in excluding a diagnosis of HIT but will result in overdiagnosis of HIT in situations where thrombocytopenia and thrombosis due to other etiologies are common, such as in the intensive care unit. A scoring model based on broad expert opinion (the HIT Expert Probability [HEP] Score) has improved operating characteristics and may provide better utility as a scoring system.

FIGURE 140-3 Time course of heparin-induced thrombocytopenia (HIT) development after heparin exposure. The timing of development after heparin exposure is a critical factor in determining the likelihood of HIT in a patient. HIT occurs early after heparin exposure in the presence of preexisting heparin/platelet factor 4 (PF4) antibodies, which disappear from circulation by ~100 days following a prior exposure. Rarely, HIT may occur later after heparin exposure (termed delayed-onset HIT). In this setting, heparin/PF4 antibody testing is usually markedly positive. HIT can occur after exposure to either unfractionated (UFH) or low-molecular-weight heparin (LMWH).
LABORATORY TESTING FOR HIT HIT (anti-heparin/PF4) antibodies can be detected using two types of assays. The most widely available is an enzyme-linked immunoassay (ELISA) with PF4/polyanion complex as the antigen. Because many patients develop antibodies but do not develop clinical HIT, the test has a low specificity for the diagnosis of HIT. This is especially true in patients who have undergone cardiopulmonary bypass surgery, where approximately 50% of patients develop these antibodies postoperatively. IgG-specific ELISAs increase specificity but may decrease sensitivity. The other assay is a platelet activation assay, most commonly the serotonin release assay, which measures the ability of the patient’s serum to activate platelets in the presence of heparin in a concentration-dependent manner. This test has lower sensitivity but higher specificity than the ELISA. However, HIT remains a clinical diagnosis.
Immune Thrombocytopenic Purpura Immune thrombocytopenic purpura (ITP; also termed idiopathic thrombocytopenic purpura) is an acquired disorder in which there is immune-mediated destruction of platelets and possibly inhibition of platelet release from the megakaryocyte. In children, it is usually an acute disease, most commonly following an infection, and with a self-limited course. In adults, it is a more chronic disease, although in some adults, spontaneous remission occurs, usually within months of diagnosis. ITP is termed secondary if it is associated with an underlying disorder; autoimmune disorders, particularly systemic lupus erythematosus (SLE), and infections, such as HIV and hepatitis C, are common causes. The association of ITP with Helicobacter pylori infection is unclear.
ITP is characterized by mucocutaneous bleeding and a low, often very low, platelet count, with an otherwise normal peripheral blood cells and smear. Patients usually present either with ecchymoses and petechiae, or with thrombocytopenia incidentally found on a routine CBC. Mucocutaneous bleeding, such as oral mucosa, gastrointestinal, or heavy menstrual bleeding, may be present. Rarely, life-threatening, including central nervous system, bleeding can occur. Wet purpura (blood blisters in the mouth) and retinal hemorrhages may herald life-threatening bleeding.
LABORATORY TESTING IN ITP Laboratory testing for antibodies (serologic testing) is usually not helpful due to the low sensitivity and specificity of the current tests. Bone marrow examination can be reserved for those who have other signs or laboratory abnormalities not explained by ITP or in patients who do not respond to initial therapy. The peripheral blood smear may show large platelets, with otherwise normal morphology. Depending on the bleeding history, iron-deficiency anemia may be present.
Laboratory testing is performed to evaluate for secondary causes of ITP and should include testing for HIV infection and hepatitis C (and other infections if indicated). Serologic testing for SLE, serum protein electrophoresis, immunoglobulin levels to potentially detect hypogammaglobulinemia, selective testing for IgA deficiency or monoclonal gammopathies, and testing for H. pylori infection should be considered, depending on the clinical circumstance. If anemia is present, direct antiglobulin testing (Coombs’ test) should be performed to rule out combined autoimmune hemolytic anemia with ITP (Evans’ syndrome).
Inherited Thrombocytopenia Thrombocytopenia is rarely inherited, either as an isolated finding or as part of a syndrome, and may be inherited in an autosomal dominant, autosomal recessive, or X-linked pattern. Many forms of autosomal dominant thrombocytopenia are now known to be associated with mutations in the nonmuscle myosin heavy chain MYH9 gene. Interestingly, these include the May-Hegglin anomaly, and Sebastian, Epstein’s, and Fechtner syndromes, all of which have distinct distinguishing features. A common feature of these disorders is large platelets (Fig. 140-1C). Autosomal recessive disorders include congenital amegakaryocytic thrombocytopenia, thrombocytopenia with absent radii, and Bernard-Soulier syndrome. The latter is primarily a functional platelet disorder due to absence of Gp Ib-IX-V, the VWF adhesion receptor. X-linked disorders include Wiskott-Aldrich syndrome and a dyshematopoietic syndrome resulting from a mutation in GATA-1, an important transcriptional regulator of hematopoiesis.
THROMBOTIC THROMBOCYTOPENIC PURPURA AND HEMOLYTIC-UREMIC SYNDROME
Thrombotic thrombocytopenic microangiopathies are a group of disorders characterized by thrombocytopenia, a microangiopathic hemolytic anemia evident by fragmented RBCs (Fig. 140-1D) and laboratory evidence of hemolysis, and microvascular thrombosis. They include thrombotic thrombocytopenic purpura (TTP) and hemolytic-uremic syndrome (HUS), as well as syndromes complicating bone marrow transplantation, certain medications and infections, pregnancy, and vasculitis. In DIC, although thrombocytopenia and microangiopathy are seen, a coagulopathy predominates, with consumption of clotting factors and fibrinogen resulting in an elevated prothrombin time (PT) and often activated partial thromboplastin time (aPTT). The PT and aPTT are characteristically normal in TTP or HUS.
Thrombotic Thrombocytopenic Purpura TTP and HUS were previously considered overlap syndromes. However, in the past few years, the pathophysiology of inherited and idiopathic TTP has become better understood and clearly differs from HUS. TTP was first described in 1924 by Eli Moschcowitz and characterized by a pentad of findings that include microangiopathic hemolytic anemia, thrombocytopenia, renal failure, neurologic findings, and fever. The full-blown syndrome is less commonly seen now, probably due to earlier diagnosis. The introduction of treatment with plasma exchange markedly improved the prognosis in patients, with a decrease in mortality from 85–100% to 10–30%.
The pathogenesis of inherited (Upshaw-Schulman syndrome) and idiopathic TTP is related to a deficiency of, or antibodies to, the metalloprotease ADAMTS13, which cleaves VWF. VWF is normally secreted as ultra-large multimers, which are then cleaved by ADAMTS13. The persistence of ultra-large VWF molecules is thought to contribute to pathogenic platelet adhesion and aggregation (Fig. 140-4). This defect alone, however, is not sufficient to result in TTP because individuals with a congenital absence of ADAMTS13 develop TTP only episodically. Additional provocative factors have not been defined. The level of ADAMTS13 activity, as well as antibodies, can now be detected by laboratory assays. Although assays with sufficient sensitivity and specificity to direct clinical management have yet to be clearly defined, ADAMTS13 activity levels of <10% are more clearly associated with idiopathic TTP.
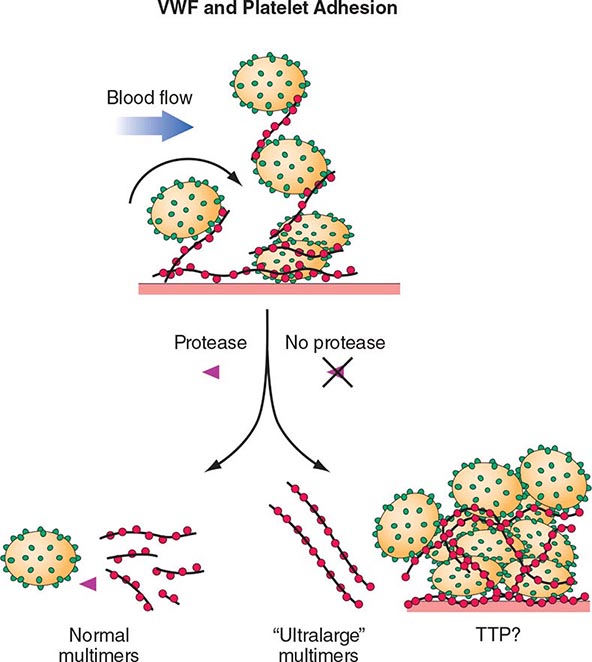
FIGURE 140-4 Pathogenesis of thrombotic thrombocytopenic purpura (TTP). Normally the ultra-high-molecular-weight multimers of von Willebrand factor (VWF) produced by the endothelial cells are processed into smaller multimers by a plasma metalloproteinase called ADAMTS13. In TTP, the activity of the protease is inhibited, and the ultra-high-molecular-weight multimers of VWF initiate platelet aggregation and thrombosis.
Idiopathic TTP appears to be more common in women than in men. No geographic or racial distribution has been defined. TTP is more common in patients with HIV infection and in pregnant women. TTP in pregnancy is not clearly related to ADAMTS13. Medication-related microangiopathic hemolytic anemia may be secondary to antibody formation (ticlopidine and possibly clopidogrel) or direct endothelial toxicity (cyclosporine, mitomycin C, tacrolimus, quinine), although this is not always so clear, and fear of withholding treatment, as well as lack of other treatment alternatives, results in broad application of plasma exchange. However, withdrawal, or reduction in dose, of endothelial toxic agents usually decreases the microangiopathy.
Hemolytic-Uremic Syndrome HUS is a syndrome characterized by acute renal failure, microangiopathic hemolytic anemia, and thrombocytopenia. It is seen predominantly in children and in most cases is preceded by an episode of diarrhea, often hemorrhagic in nature. Escherichia coli O157:H7 is the most frequent, although not only, etiologic serotype. HUS not associated with diarrhea is more heterogeneous in presentation and course. Atypical HUS (aHUS) due to genetic defects that result in chronic complement activation has been defined, and screening for mutations in complement regulatory genes is available.
THROMBOCYTOSIS
Thrombocytosis is almost always due to (1) iron deficiency; (2) inflammation, cancer, or infection (reactive thrombocytosis); or (3) an underlying myeloproliferative process (essential thrombocythemia or polycythemia vera) (Chap. 131) or, rarely, the 5q– myelodysplastic process (Chap. 130). Patients presenting with an elevated platelet count should be evaluated for underlying inflammation or malignancy, and iron deficiency should be ruled out. Thrombocytosis in response to acute or chronic inflammation has not been clearly associated with an increased thrombotic risk. In fact, patients with markedly elevated platelet counts (>1.5 million), usually seen in the setting of a myeloproliferative disorder, have an increased risk of bleeding. This appears to be due, at least in part, to acquired von Willebrand disease (VWD) due to platelet-VWF binding and removal from the circulation.
QUALITATIVE DISORDERS OF PLATELET FUNCTION
Inherited Disorders of Platelet Function Inherited platelet function disorders are thought to be relatively rare, although the prevalence of mild disorders of platelet function is unclear, in part because our testing for such disorders is suboptimal. Rare qualitative disorders include the autosomal recessive disorders Glanzmann’s thrombasthenia (absence of the platelet Gp IIb/IIIa receptor) and Bernard-Soulier syndrome (absence of the platelet Gp Ib-IX-V receptor). Both are inherited in an autosomal recessive fashion and present with bleeding symptoms in childhood.
Platelet storage pool disorder (SPD) is the classic autosomal dominant qualitative platelet disorder. This results from abnormalities of platelet granule formation. It is also seen as a part of inherited disorders of granule formation, such as Hermansky-Pudlak syndrome. Bleeding symptoms in SPD are variable, but often are mild. The most common inherited disorders of platelet function prevent normal secretion of granule content and are termed secretion defects. Few of these abnormalities have been dissected at the molecular level but they likely result from various mutations..
Acquired Disorders of Platelet Function Acquired platelet dysfunction is common, usually due to medications, either intentionally as with antiplatelet therapy or unintentionally as with high-dose penicillins. Acquired platelet dysfunction occurs in uremia. This is likely multifactorial, but the resultant effect is defective adhesion and activation. The platelet defect is improved most by dialysis but may also be improved by increasing the hematocrit to 27–32%, giving DDAVP (0.3 μg/kg), or use of conjugated estrogens. Platelet dysfunction also occurs with cardiopulmonary bypass due to the effect of the artificial circuit on platelets, and bleeding symptoms respond to platelet transfusion. Platelet dysfunction seen with underlying hematologic disorders can result from nonspecific interference by circulating paraproteins or intrinsic platelet defects in myeloproliferative and myelodysplastic syndromes.
VON WILLEBRAND DISEASE
VWD is the most common inherited bleeding disorder. Estimates from laboratory data suggest a prevalence of approximately 1%, but data based on symptomatic individuals suggest that it is closer to 0.1% of the population. VWF serves two roles: (1) as the major adhesion molecule that tethers the platelet to the exposed subendothelium; and (2) as the binding protein for factor VIII (FVIII), resulting in significant prolongation of the FVIII half-life in circulation. The platelet-adhesive function of VWF is critically dependent on the presence of large VWF multimers, whereas FVIII binding is not. Most of the symptoms of VWD are “platelet-like” except in more severe VWD when the FVIII is low enough to produce symptoms similar to those found in FVIII deficiency (hemophilia A).
VWD has been classified into three major types, with four subtypes of type 2 (Table 140-2; Fig. 140-5). By far the most common type of VWD is type 1 disease, with a parallel decrease in VWF protein, VWF function, and FVIII levels, accounting for at least 80% of cases. Patients have predominantly mucosal bleeding symptoms, although postoperative bleeding can also be seen. Bleeding symptoms are very uncommon in infancy and usually manifest later in childhood with excessive bruising and epistaxis. Because these symptoms occur commonly in childhood, the clinician should particularly note bruising at sites unlikely to be traumatized and/or prolonged epistaxis requiring medical attention. Menorrhagia is a common manifestation of VWD. Menstrual bleeding resulting in anemia should warrant an evaluation for VWD and, if negative, functional platelet disorders. Frequently, mild type 1 VWD first manifests with dental extractions, particularly wisdom tooth extraction, or tonsillectomy.
LABORATORY DIAGNOSIS OF VON WILLEBRAND DISEASE (VWD) |
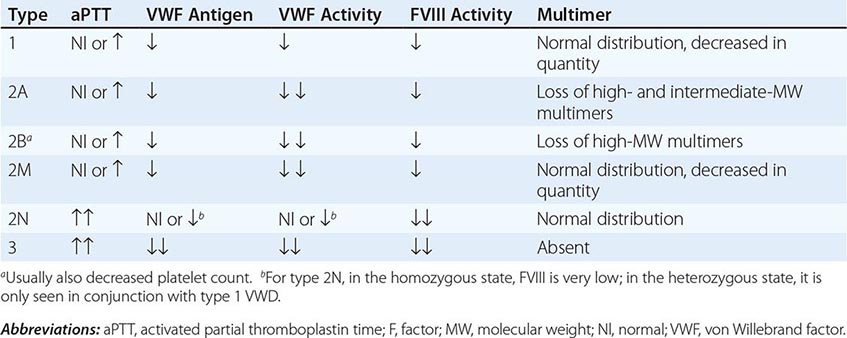
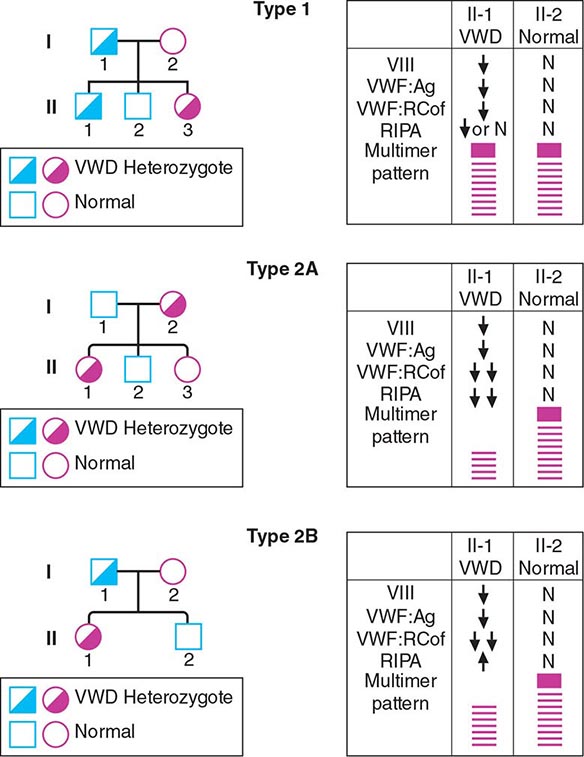
FIGURE 140-5 Pattern of inheritance and laboratory findings in von Willebrand disease (VWD). The assays of platelet function include a coagulation assay of factor VIII bound and carried by von Willebrand factor (VWF), abbreviated as VIII; immunoassay of total VWF protein (VWF:Ag); bioassay of the ability of patient plasma to support ristocetin-induced agglutination of normal platelets (VWF:RCoF); and ristocetin-induced aggregation of patient platelets, abbreviated RIPA. The multimer pattern illustrates the protein bands present when plasma is electrophoresed in a polyacrylamide gel. The II-1 and II-2 columns refer to the phenotypes of the second-generation offspring.
Not all patients with low VWF levels have bleeding symptoms. Whether patients bleed or not will depend on the overall hemostatic balance they have inherited, along with environmental influences and the type of hemostatic challenges they experience. Although the inheritance of VWD is autosomal, many factors modulate both VWF levels and bleeding symptoms. These have not all been defined, but include blood type, thyroid hormone status, race, stress, exercise, and hormonal (both endogenous and exogenous) influences. Patients with type O blood have VWF protein levels of approximately one-half that of patients with AB blood type; and, in fact, the normal range for patients with type O blood overlaps that which has been considered diagnostic for VWD. A mildly decreased VWF level should be viewed more as a risk factor for bleeding than as an actual disease.
Patients with type 2 VWD have functional defects; thus, the VWF antigen measurement is significantly higher than the test of function. For types 2A, 2B, and 2M VWD, platelet-binding and/or collagen binding VWF activity is decreased. In type 2A VWD, the impaired function is due either to increased susceptibility to cleavage by ADAMTS13, resulting in loss of intermediate- and high-molecular-weight multimers, or to decreased secretion of these multimers by the cell. Type 2B VWD results from gain-of-function mutations that result in increased spontaneous binding of VWF to platelets in circulation, with subsequent clearance of this complex by the reticuloendothelial system. The resulting VWF in the patients’ plasma lacks the highest molecular-weight multimers, and the platelet count is usually modestly reduced. Type 2M occurs as a consequence of a group of mutations that cause dysfunction but do not affect multimer structure.
Type 2N VWD is due to mutations in VWF that affect binding of FVIII. As FVIII is stabilized by binding to VWF, the FVIII in patients with type 2N VWD has a very short half-life, and the FVIII level is markedly decreased. This is sometimes termed autosomal hemophilia. Type 3 VWD, or severe VWD, describes patients with virtually no VWF protein and FVIII levels <10%. Patients experience mucosal and joint bleeding, surgery-related bleeding, and other bleeding symptoms. Some patients with type 3 VWD, particularly those with large VWF gene deletions, are at risk of developing antibodies to infused VWF.
Acquired VWD is a rare disorder, most commonly seen in patients with underlying lymphoproliferative disorders, including monoclonal gammopathies of underdetermined significance (MGUS), multiple myeloma, and Waldenström’s macroglobulinemia. It is seen most commonly in the setting of MGUS and should be suspected in patients, particularly elderly patients, with a new onset of severe mucosal bleeding symptoms. Laboratory evidence of acquired VWD is found in some patients with aortic valvular disease. Heyde’s syndrome (aortic stenosis with gastrointestinal bleeding) is attributed to the presence of angiodysplasia of the gastrointestinal tract in patients with aortic stenosis. The shear stress on blood passing through the stenotic aortic valve appears to produce a change in VWF, making it susceptible to serum proteases. Consequently, large multimer forms are lost, leading to an acquired type 2 VWD, but return when the stenotic valve is replaced.
DISORDERS OF THE VESSEL WALL
The vessel wall is an integral part of hemostasis, and separation of a fluid phase is artificial, particularly in disorders such as TTP or HIT that clearly involve the endothelium as well. Inflammation localized to the vessel wall, such as vasculitis, and inherited connective tissue disorders are abnormalities inherent to the vessel wall.
METABOLIC AND INFLAMMATORY DISORDERS Acute febrile illnesses may result in vascular damage. This can result from immune complexes containing viral antigens or the viruses themselves. Certain pathogens, such as the rickettsiae causing Rocky Mountain spotted fever, replicate in endothelial cells and damage them. Vascular purpura may occur in patients with polyclonal gammopathies but more commonly in those with monoclonal gammopathies, including Waldenström’s macroglobulinemia, multiple myeloma, and cryoglobulinemia. Patients with mixed cryoglobulinemia develop a more extensive maculopapular rash due to immune complex–mediated damage to the vessel wall.
Patients with scurvy (vitamin C deficiency) develop painful episodes of perifollicular skin bleeding as well as more systemic bleeding symptoms. Vitamin C is needed to synthesize hydroxyproline, an essential constituent of collagen. Patients with Cushing’s syndrome or on chronic glucocorticoid therapy develop skin bleeding and easy bruising due to atrophy of supporting connective tissue. A similar phenomenon is seen with aging, where following minor trauma, blood spreads superficially under the epidermis. This has been termed senile purpura. It is most common on skin that has been previously damaged by sun exposure.
Henoch-Schönlein, or anaphylactoid, purpura is a distinct, self-limited type of vasculitis that occurs in children and young adults. Patients have an acute inflammatory reaction with IgA and complement components in capillaries, mesangial tissues, and small arterioles leading to increased vascular permeability and localized hemorrhage. The syndrome is often preceded by an upper respiratory infection, commonly with streptococcal pharyngitis, or is triggered by drug or food allergies. Patients develop a purpuric rash on the extensor surfaces of the arms and legs, usually accompanied by polyarthralgias or arthritis, abdominal pain, and hematuria from focal glomerulonephritis. All coagulation tests are normal, but renal impairment may occur. Glucocorticoids can provide symptomatic relief but do not alter the course of the illness.
INHERITED DISORDERS OF THE VESSEL WALL Patients with inherited disorders of the connective tissue matrix, such as Marfan’s syndrome, Ehlers-Danlos syndrome, and pseudoxanthoma elasticum, frequently report easy bruising. Inherited vascular abnormalities can result in increased bleeding. This is notably seen in hereditary hemorrhagic telangiectasia (HHT, or Osler-Weber-Rendu disease), a disorder where abnormal telangiectatic capillaries result in frequent bleeding episodes, primarily from the nose and gastrointestinal tract. Arteriovenous malformation (AVM) in the lung, brain, and liver may also occur in HHT. The telangiectasia can often be visualized on the oral and nasal mucosa. Signs and symptoms develop over time. Epistaxis begins, on average, at the age of 12 and occurs in >95% of affected individuals by middle age. Two genes involved in the pathogenesis are eng (endoglin) on chromosome 9q33-34 (so-called HHT type 1), associated with pulmonary AVM in 40% of cases, and alk1 (activin-receptor-like kinase 1) on chromosome 12q13, associated with a much lower risk of pulmonary AVM.
ACKNOWLEDGMENT
Robert Handin, MD, contributed this chapter in the 16th edition and some materials from his chapter are included here.
141 | Coagulation Disorders |
Deficiencies of coagulation factors have been recognized for centuries. Patients with genetic deficiencies of plasma coagulation factors exhibit life-long recurrent bleeding episodes into joints, muscles, and closed spaces, either spontaneously or following an injury. The most common inherited factor deficiencies are the hemophilias, X-linked diseases caused by deficiency of factor (F) VIII (hemophilia A) or FIX (hemophilia B). Rare congenital bleeding disorders due to deficiencies of other factors, including FII (prothrombin), FV, FVII, FX, FXI, and FXIII, and fibrinogen are commonly inherited in an autosomal recessive manner (Table 141-1). Advances in characterization of the molecular bases of clotting factor deficiencies have contributed to better understanding of the disease phenotypes and may eventually allow more targeted therapeutic approaches through the development of small molecules, recombinant proteins, or cell and gene-based therapies.
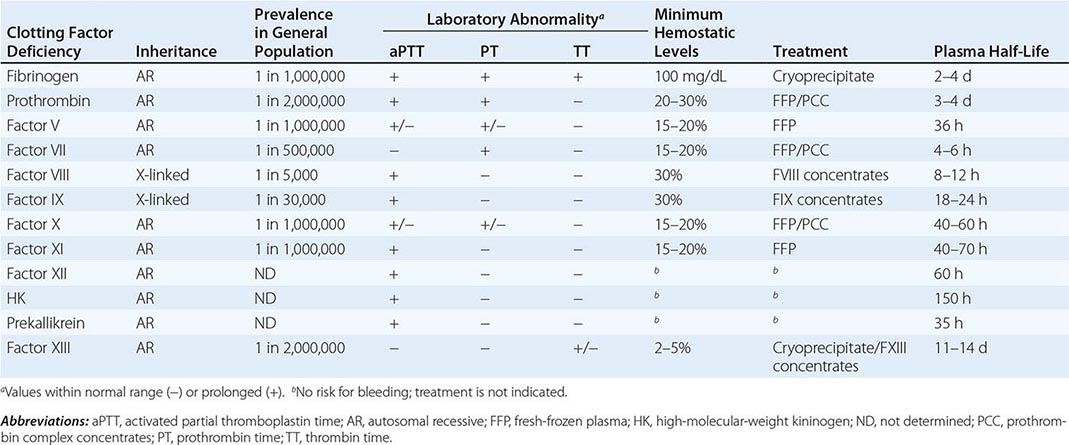
GENETIC AND LABORATORY CHARACTERISTICS OF INHERITED COAGULATION DISORDERS |
Commonly used tests of hemostasis provide the initial screening for clotting factor activity (Fig. 141-1), and disease phenotype often correlates with the level of clotting activity. An isolated abnormal prothrombin time (PT) suggests FVII deficiency, whereas a prolonged activated partial thromboplastin time (aPTT) indicates most commonly hemophilia or FXI deficiency (Fig. 141-1). The prolongation of both PT and aPTT suggests deficiency of FV, FX, FII, or fibrinogen abnormalities. The addition of the missing factor at a range of doses to the subject’s plasma will correct the abnormal clotting times; the result is expressed as a percentage of the activity observed in normal subjects.
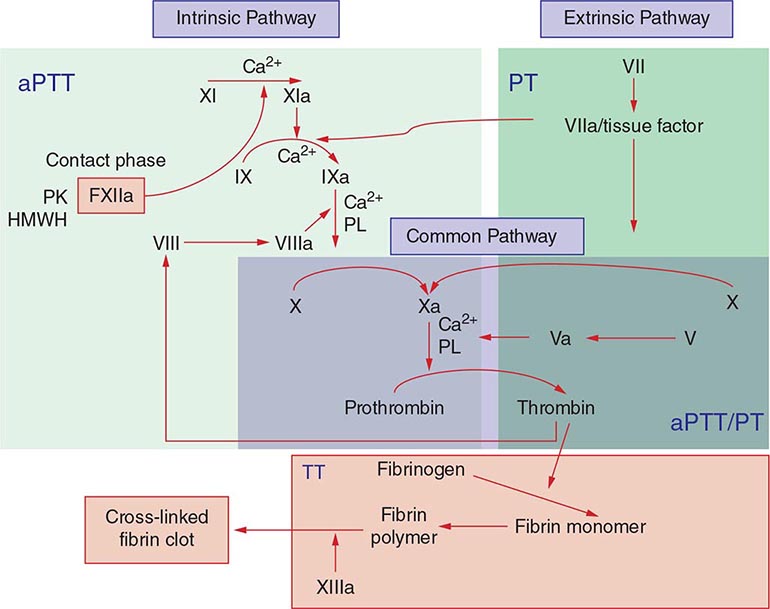
FIGURE 141-1 Coagulation cascade and laboratory assessment of clotting factor deficiency by activated partial prothrombin time (aPTT), prothrombin time (PT), thrombin time (TT), and phospholipid (PL).
Acquired deficiencies of plasma coagulation factors are more frequent than congenital disorders; the most common disorders include hemorrhagic diathesis of liver disease, disseminated intravascular coagulation (DIC), and vitamin K deficiency. In these disorders, blood coagulation is hampered by the deficiency of more than one clotting factor, and the bleeding episodes are the result of perturbation of both primary (coagulation) and secondary (e.g., platelet and vessel wall interactions) hemostasis.
The development of antibodies to coagulation plasma proteins, clinically termed inhibitors, is a relatively rare disease that often affects hemophilia A or B and FXI-deficient patients on repetitive exposure to the missing protein to control bleeding episodes. Inhibitors also occur among subjects without genetic deficiency of clotting factors (e.g., in the postpartum setting as a manifestation of underlying autoimmune or neoplastic disease or idiopathically). Rare cases of inhibitors to thrombin or FV have been reported in patients receiving topical bovine thrombin preparation as a local hemostatic agent in complex surgeries. The diagnosis of inhibitors is based on the same tests as those used to diagnose inherited plasma coagulation factor deficiencies. However, the addition of the missing protein to the plasma of a subject with an inhibitor does not correct the abnormal aPTT and/or PT tests (known as mixing tests). This is the major laboratory difference between deficiencies and inhibitors. Additional tests are required to measure the specificity of the inhibitor and its titer.
The treatment of these bleeding disorders often requires replacement of the deficient protein using recombinant or purified plasma-derived products or fresh-frozen plasma (FFP). Therefore, it is imperative to arrive at a proper diagnosis to optimize patient care without unnecessary exposure to suboptimal treatment and the risks of bloodborne disease.
HEMOPHILIA
PATHOGENESIS AND CLINICAL MANIFESTATIONS
Hemophilia is an X-linked recessive hemorrhagic disease due to mutations in the F8 gene (hemophilia A or classic hemophilia) or F9 gene (hemophilia B). The disease affects 1 in 10,000 males worldwide, in all ethnic groups; hemophilia A represents 80% of all cases. Male subjects are clinically affected; women, who carry a single mutated gene, are generally asymptomatic. Family history of the disease is absent in ~30% of cases, and in these cases, 80% of the mothers are carriers of the de novo mutated allele. More than 500 different mutations have been identified in the F8 or F9 genes of patients with hemophilia A or B, respectively. One of the most common hemophilia A mutations results from an inversion of the intron 22 sequence, and it is present in 40% of cases of severe hemophilia A. Advances in molecular diagnosis now permit precise identification of mutations, allowing accurate diagnosis of women carriers of the hemophilia gene in affected families.
Clinically, hemophilia A and hemophilia B are indistinguishable. The disease phenotype correlates with the residual activity of FVIII or FIX and can be classified as severe (<1%), moderate (1–5%), or mild (6–30%). In the severe and moderate forms, the disease is characterized by bleeding into the joints (hemarthrosis), soft tissues, and muscles after minor trauma or even spontaneously. Patients with mild disease experience infrequent bleeding that is usually secondary to trauma. Among those with residual FVIII or FIX activity >25% of normal, the disease is discovered only by bleeding after major trauma or during routine presurgery laboratory tests. Typically, the global tests of coagulation show only an isolated prolongation of the aPTT assay. Patients with hemophilia have normal bleeding times and platelet counts. The diagnosis is made after specific determination of FVIII or FIX clotting activity.
Early in life, bleeding may present after circumcision or rarely as intracranial hemorrhages. The disease is more evident when children begin to walk or crawl. In the severe form, the most common bleeding manifestations are the recurrent hemarthroses, which can affect every joint but mainly affect knees, elbows, ankles, shoulders, and hips. Acute hemarthroses are painful, and clinical signs are local swelling and erythema. To avoid pain, the patient may adopt a fixed position, which leads eventually to muscle contractures. Very young children unable to communicate verbally show irritability and a lack of movement of the affected joint. Chronic hemarthroses are debilitating, with synovial thickening and synovitis in response to the intraarticular blood. After a joint has been damaged, recurrent bleeding episodes result in the clinically recognized “target joint,” which then establishes a vicious cycle of bleeding, resulting in progressive joint deformity that in critical cases requires surgery as the only therapeutic option. Hematomas into the muscle of distal parts of the limbs may lead to external compression of arteries, veins, or nerves that can evolve to a compartment syndrome.
Bleeding into the oropharyngeal spaces, central nervous system (CNS), or retroperitoneum is life threatening and requires immediate therapy. Retroperitoneal hemorrhages can accumulate large quantities of blood with formation of masses with calcification and inflammatory tissue reaction (pseudotumor syndrome) and also result in damage to the femoral nerve. Pseudotumors can also form in bones, especially long bones of the lower limbs. Hematuria is frequent among hemophilia patients, even in the absence of genitourinary pathology. It is often self-limited and may not require specific therapy.
HEMOPHILIA |
Without treatment, severe hemophilia has a limited life expectancy. Advances in the blood fractionation industry during World War II resulted in the realization that plasma could be used to treat hemophilia, but the volumes required to achieve even modest elevation of circulating factor levels limit the utility of plasma infusion as an approach to disease management. The discovery in the 1960s that the cryoprecipitate fraction of plasma was enriched for FVIII, and the eventual purification of FVIII and FIX from plasma, led to the introduction of home infusion therapy with factor concentrates in the 1970s. The availability of factor concentrates resulted in a dramatic improvement in life expectancy and in quality of life for people with severe hemophilia. However, the contamination of the blood supply with hepatitis viruses and, subsequently, HIV resulted in widespread transmission of these bloodborne infections within the hemophilia population; complications of HIV and of hepatitis C are now the leading causes of death among U.S. adults with severe hemophilia. The introduction of viral inactivation steps in the preparation of plasma-derived products in the mid-1980s greatly reduced the risk of HIV and hepatitis, and the risks were further reduced by the successful production of recombinant FVIII and FIX proteins, both licensed in the 1990s. It is uncommon for hemophilic patients born after 1985 to have contracted either hepatitis or HIV, and for these individuals, life expectancy is approximately 65 years. In fact, since 1998, no evidence of new infections with viral hepatitis or HIV has been reported in patients using blood products. Factor replacement therapy for hemophilia can be provided either in response to a bleeding episode or as a prophylactic treatment. Primary prophylaxis is defined as a strategy for maintaining the missing clotting factor at levels ~1% or higher on a regular basis in order to prevent bleeds, especially the onset of hemarthroses. Hemophilic boys receiving regular infusions of FVIII (3 days/week) or FIX (2 days/week) can reach puberty without detectable joint abnormalities. Prophylaxis has become gradually more common in young patients. The Centers for Disease Control and Prevention reported that 51% of children with severe hemophilia who are younger than age 6 years receive prophylaxis, increasing considerably from 33% in 1995. Although highly recommended, the high cost and difficulties in accessing peripheral veins in young patients and the potential infectious and thrombotic risks of long-term central vein catheters are important limiting factors for many young patients. Emerging data show that prophylaxis is also increasing among adults with severe hemophilia.
General considerations regarding the treatment of bleeds in hemophilia include the following: (1) Treatment should begin as soon as possible because symptoms often precede objective evidence of bleeding; because of the superior efficacy of early therapeutic intervention, classic symptoms of bleeding into the joint in a reliable patient, headaches, or automobile or other accidents require prompt replacement and further laboratory investigation. (2) Drugs that hamper platelet function, such as aspirin or aspirin- containing drugs, should be avoided; to control pain, drugs such as ibuprofen or propoxyphene are preferred. FVIII and FIX are dosed in units. One unit is defined as amount of FVIII (100 ng/mL) or FIX (5 μg/mL) in 1 mL of normal plasma. One unit of FVIII per kilogram of body weight increases the plasma FVIII level by 2%. One can calculate the dose needed to increase FVIII levels to 100% in a 70-kg severe hemophilia patient (<1%) using the simple formula below. Thus, 3500 units of FVIII will raise the circulating level to 100%.
![]()
The doses for FIX replacement are different from those for FVIII, because FIX recovery after infusion is usually only 50% of the predicted value. Therefore, the formula for FIX replacement is as follows:
![]()
The FVIII half-life of 8–12 h requires injections twice a day to maintain therapeutic levels, whereas the FIX half-life is longer, ~24 h, so that once-a-day injection is sufficient. In specific situations such as after surgery, continuous infusion of factor may be desirable because of its safety in achieving sustained factor levels at a lower total cost.
Cryoprecipitate is enriched with FVIII protein (each bag contains ~80 IU of FVIII) and was commonly used for the treatment of hemophilia A decades ago; it is still in use in some developing countries, but because of the risk of bloodborne diseases, this product should be avoided in hemophilia patients when factor concentrates are available.
Mild bleeds such as uncomplicated hemarthroses or superficial hematomas require initial therapy with factor levels of 30–50%. Additional doses to maintain levels of 15–25% for 2 or 3 days are indicated for severe hemarthroses, especially when these episodes affect the “target joint.” Large hematomas, or bleeds into deep muscles, require factor levels of 50% or even higher if the clinical symptoms do not improve, and factor replacement may be required for a period of 1 week or longer. The control of serious bleeds including those that affect the oropharyngeal spaces, CNS, and the retroperitoneum require sustained protein levels of 50–100% for 7–10 days. Prophylactic replacement for surgery is aimed at achieving normal factor levels (100%) for a period of 7–10 days; replacement can then be tapered depending on the extent of the surgical wounds. Oral surgery is associated with extensive tissue damage that usually requires factor replacement for 1–3 days coupled with oral antifibrinolytic drugs.
NONTRANSFUSION THERAPY IN HEMOPHILIA
DDAVP (1-Amino-8-D-Arginine Vasopressin) DDAVP is a synthetic vasopressin analog that causes a transient rise in FVIII and von Willebrand factor (VWF), but not FIX, through a mechanism involving release from endothelial cells. Patients with moderate or mild hemophilia A should be tested to determine if they respond to DDAVP before a therapeutic application. DDAVP at doses of 0.3 μg/kg body weight, over a 20-min period, is expected to raise FVIII levels by two- to threefold over baseline, peaking between 30 and 60 min after infusion. DDAVP does not improve FVIII levels in severe hemophilia A patients, because there are no stores to release. Repeated dosing of DDAVP results in tachyphylaxis because the mechanism is an increase in release rather than de novo synthesis of FVIII and VWF. More than three consecutive doses become ineffective, and if further therapy is indicated, FVIII replacement is required to achieve hemostasis.
Antifibrinolytic Drugs Bleeding in the gums, gastrointestinal tract, and during oral surgery requires the use of oral antifibrinolytic drugs such as ε-amino caproic acid (EACA) or tranexamic acid to control local hemostasis. The duration of the treatment depending on the clinical indication is 1 week or longer. Tranexamic acid is given at doses of 25 mg/kg three to four times a day. EACA treatment requires a loading dose of 200 mg/kg (maximum of 10 g) followed by 100 mg/kg per dose (maximum 30 g/d) every 6 h. These drugs are not indicated to control hematuria because of the risk of formation of an occlusive clot in the lumen of genitourinary tract structures.
COMPLICATIONS
Inhibitor Formation The formation of alloantibodies to FVIII or FIX is currently the major complication of hemophilia treatment. The prevalence of inhibitors to FVIII is estimated to be between 5 and 10% of all cases and ~20% of severe hemophilia A patients. Inhibitors to FIX are detected in only 3–5% of all hemophilia B patients. The high-risk group for inhibitor formation includes severe deficiency (>80% of all cases of inhibitors), familial history of inhibitor, African descent, mutations in the FVIII or FIX gene resulting in deletion of large coding regions, or gross gene rearrangements. Inhibitors usually appear early in life, at a median of 2 years of age, and after 10 cumulative days of exposure. However, intensive replacement therapy such as for major surgery, intracranial bleeding, or trauma increases the risk of inhibitor formation for patients of all ages and degree of clinical severity, which requires close laboratory monitoring in the following weeks.
The clinical diagnosis of an inhibitor is suspected when patients do not respond to factor replacement at therapeutic doses. Inhibitors increase both morbidity and mortality in hemophilia. Because early detection of an inhibitor is critical to a successful correction of the bleeding or to eradication of the antibody, most hemophilia centers perform annual screening for inhibitors. The laboratory test required to confirm the presence of an inhibitor is an aPTT with a mix (with normal plasma). In most hemophilia patients, a 1:1 mix with normal plasma completely corrects the aPTT. In inhibitor patients, the aPTT on a 1:1 mix is abnormally prolonged, because the inhibitor neutralizes the FVIII clotting activity of the normal plasma. The Bethesda assay uses a similar principle and defines the specificity of the inhibitor and its titer. The results are expressed in Bethesda units (BU), in which 1 BU is the amount of antibody that neutralizes 50% of the FVIII or FIX present in normal plasma after 2 h of incubation at 37°C. Clinically, inhibitor patients are classified as low responders or high responders, which provides guidelines for optimal therapy. Therapy for inhibitor patients has two goals: the control of acute bleeding episodes and the eradication of the inhibitor. For the control of bleeding episodes, low responders, those with titer <5 BU, respond well to high doses of human or porcine FVIII (50–100 U/kg), with minimal or no increase in the inhibitor titers. However, high-responder patients, those with initial inhibitor titer >10 BU or an anamnestic response in the antibody titer to >10 BU even if low titer initially, do not respond to FVIII or FIX concentrates. The control of bleeding episodes in high-responder patients can be achieved by using concentrates enriched for prothrombin, FVII, FIX, FX (prothrombin complex concentrates [PCCs] or activated PCCs [aPCCs]), and more recently recombinant activated factor VII (FVIIa) known as “bypass agents” (Fig. 141-1). The rates of therapeutic success have been higher for FVIIa than for PCC or aPCC. For eradication of the inhibitory antibody, immunosuppression alone is not effective. The most effective strategy is the immune tolerance induction (ITI) based on daily infusion of missing protein until the inhibitor disappears, typically requiring periods longer than 1 year, with success rates of approximately 60%. The management of patients with severe hemophilia and inhibitors resistant to ITI is challenging. The use of anti-CD20 monoclonal antibody (rituximab) combined with ITI was thought to be effective. Although this therapy may reduce the inhibitor titers in some cases, sustained eradication is uncommon and may require two to three infusions weekly of clotting factor concentrates.
Novel Therapeutic Approaches in Development for Hemophilia Clinical studies using long-acting clotting factors with prolonged half-lives are in the late phase of clinical testing, and these new-generation products (for FVIII and FIX) may facilitate prophylaxis by requiring fewer injections to maintain circulating levels above 1%.
The use of recombinant interleukin 11 in patients with moderate or mild hemophilia A unresponsive to DDAVP has been tested in early-phase clinical trials and may be an alternate therapeutic strategy for clinical situations that require transient increases in FVIII levels.
Gene therapy trials for hemophilia B using adeno-associated viral vectors are ongoing, and initial data are promising (Chap. 91e).
INFECTIOUS DISEASES
Hepatitis C virus (HCV) infection is the major cause of morbidity and the second leading cause of death in hemophilia patients exposed to older clotting factor concentrates. The vast majority of young patients treated with plasma-derived products from 1970 to 1985 became infected with HCV. It has been estimated that >80% of patients older than 20 years of age are HCV antibody positive as of 2006. The comorbidity of the underlying liver disease in hemophilia patients is clear when these individuals require invasive procedures; correction of both genetic and acquired (secondary to liver disease) deficiencies may be needed. Infection with HIV also swept the population of patients using plasma-derived concentrates two decades ago. Co-infection of HCV and HIV, present in almost 50% of hemophilia patients, is an aggravating factor for the evolution of liver disease. The response to HCV antiviral therapy in hemophilia is restricted to <30% of patients and even poorer among those with both HCV and HIV infection. End-stage liver disease requiring organ transplantation may be curative for both the liver disease and for hemophilia.
EMERGING CLINICAL PROBLEMS IN AGING HEMOPHILIA PATIENTS
There has been continuous improvement of the management of hemophilia since the increase in the population of adults living beyond middle age in the developing world. The life expectancy of a patient with severe hemophilia is only ~10 years shorter than the general male population. In patients with mild or moderate hemophilia, life expectancy is approaching that of the male population without coagulopathy. Elderly hemophilia patients have different problems compared to the younger generation; they have more severe arthropathy and chronic pain, due to suboptimal treatment, and high rates of HCV and/or HIV infections.
Early data indicate that mortality from coronary artery disease is lower in hemophilia patients than the general male population. The underlying hypocoagulability probably provides a protective effect against thrombus formation, but it does not prevent atherogenesis. Similar to the general population, these patients are exposed to cardiovascular risk factors such as age, obesity, and smoking. Moreover, physical inactivity, hypertension, and chronic renal disease are commonly observed in hemophilia patients. In HIV patients on combined antiretroviral therapy, there may be a further increase in the risk of cardiovascular disease. Therefore, these patients should be carefully considered for preventive and therapeutic approaches to minimize the risk of cardiovascular disease.
Excessive replacement therapy should be avoided, and it is prudent to slowly infuse factor concentrates. Continuous infusion of clotting factor is preferable to bolus dosing in patients with cardiovascular risk factors undergoing invasive procedures. The management of an acute ischemic event and coronary revascularization should include the collaboration of hematologists and internists. The early assumption that hemophilia would protect against occlusive vascular disease may change in this aging population. Cancer is a common cause of mortality in aging hemophilia patients because they are at risk for HIV- and HCV-related malignancies. Hepatocellular carcinoma (HCC) is the most prevalent primary liver cancer and a common cause of death in HIV-negative patients. The recommendations for cancer screening for the general population should be the same for age-matched hemophilia patients. Among those with high-risk HCV, a semiannual or annual ultrasound and α fetoprotein are recommended for HCC. Screening for urogenital neoplasm in the presence of hematuria or hematochezia may be delayed due to the underlying bleeding disease, thus preventing early intervention. Multidisciplinary interaction should facilitate the attempts to ensure optimal cancer prevention and treatment recommendations for those with hemophilia.
MANAGEMENT OF CARRIERS OF HEMOPHILIA
Usually hemophilia carriers, with factor levels of ~50% of normal, have not been considered to be at risk for bleeding. However, a wide range of values (22–116%) have been reported due to random inactivation of the × chromosomes (lyonization). Therefore, it is important to measure the factor level of carriers to recognize those at risk of bleeding and to optimize preoperative and postoperative management. During pregnancy, both FVIII and FIX levels increase gradually until delivery. FVIII levels increase approximately two- to threefold compared to nonpregnant women, whereas an FIX increase is less pronounced. After delivery, there is a rapid fall in the pregnancy-induced rise of maternal clotting factor levels. This represents an imminent risk of bleeding that can be prevented by infusion of factor concentrate to levels of 50–70% for 3 days in the setting of vaginal delivery and up to 5 days for cesarean section. In mild cases, the use of DDAVP and/or antifibrinolytic drugs is recommended.
FACTOR XI DEFICIENCY
Factor XI is a zymogen of an active serine protease (FIXa) in the intrinsic pathway of blood coagulation that activates FIX (Fig. 141-1). There are two pathways for the formation of FXIa. In an aPTT-based assay, the protease is the result of activation by FXIIa in conjunction with high-molecular-weight kininogen and kallikrein. In vivo data suggest that thrombin is the physiologic activator of FXI. The generation of thrombin by the tissue factor/factor VIIa pathway activates FXI on the platelet surface that contributes to additional thrombin generation after the clot has formed and thus augments resistance to fibrinolysis through a thrombin-activated fibrinolytic inhibitor (TAFI).
Factor XI deficiency is a rare bleeding disorder that occurs in the general population at a frequency of one in a million. However, the disease is highly prevalent among Ashkenazi and Iraqi Jewish populations, reaching a frequency of 6% as heterozygotes and 0.1–0.3% as homozygotes. More than 65 mutations in the FXI gene have been reported, whereas fewer mutations (two to three) are found among affected Jewish populations.
Normal FXI clotting activity levels range from 70 to 150 U/dL. In heterozygous patients with moderate deficiency, FXI ranges from 20 to 70 U/dL, whereas in homozygous or double heterozygote patients, FXI levels are <1–20 U/dL. Patients with FXI levels <10% of normal have a high risk of bleeding, but the disease phenotype does not always correlate with residual FXI clotting activity. A family history is indicative of the risk of bleeding in the propositus. Clinically, the presence of mucocutaneous hemorrhages such as bruises, gum bleeding, epistaxis, hematuria, and menorrhagia are common, especially following trauma. This hemorrhagic phenotype suggests that tissues rich in fibrinolytic activity are more susceptible to FXI deficiency. Postoperative bleeding is common but not always present, even among patients with very low FXI levels.
FXI replacement is indicated in patients with severe disease required to undergo a surgical procedure. A negative history of bleeding complications following invasive procedures does not exclude the possibility of an increased risk for hemorrhage.
RARE BLEEDING DISORDERS
Collectively, the inherited disorders resulting from deficiencies of clotting factors other than FVIII, FIX, and FXI (Table 141-1) represent a group of rare bleeding diseases. The bleeding symptoms in these patients vary from asymptomatic (dysfibrinogenemia or FVII deficiency) to life-threatening (FX or FXIII deficiency). There is no pathognomonic clinical manifestation that suggests one specific disease, but overall, in contrast to hemophilia, hemarthrosis is a rare event and bleeding in the mucosal tract or after umbilical cord clamping is common. Individuals heterozygous for plasma coagulation deficiencies are often asymptomatic. The laboratory assessment for the specific deficient factor following screening with general coagulation tests (Table 141-1) will define the diagnosis.
Replacement therapy using FFP or prothrombin complex concentrates (containing prothrombin, FVII, FIX, and FX) provides adequate hemostasis in response to bleeds or as prophylactic treatment. The use of PCC should be carefully monitored and avoided in patients with underlying liver disease, or those at high risk for thrombosis because of the risk of DIC.
FAMILIAL MULTIPLE COAGULATION DEFICIENCIES
There are several bleeding disorders characterized by the inherited deficiency of more than one plasma coagulation factor. To date, the genetic defects in two of these diseases have been characterized, and they provide new insights into the regulation of hemostasis by gene-encoding proteins outside blood coagulation.
Combined Deficiency of FV and FVIII Patients with combined FV and FVIII deficiency exhibit ~5% of residual clotting activity of each factor. Interestingly, the disease phenotype is a mild bleeding tendency, often following trauma. An underlying mutation has been identified in the endoplasmic reticulum/Golgi intermediate compartment (ERGIC-53) gene, a mannose-binding protein localized in the Golgi apparatus that functions as a chaperone for both FV and FVIII. In other families, mutations in the multiple coagulation factor deficiency 2 (MCFD2) gene have been defined; this gene encodes a protein that forms a Ca2+ -dependent complex with ERGIC-53 and provides cofactor activity in the intracellular mobilization of both FV and FVIII.
Multiple Deficiencies of Vitamin K–Dependent Coagulation Factors Two enzymes involved in vitamin K metabolism have been associated with combined deficiency of all vitamin K–dependent proteins, including the procoagulant proteins prothrombin, VII, IX, and × and the anticoagulant proteins C and S. Vitamin K is a fat-soluble vitamin that is a cofactor for carboxylation of the gamma carbon of the glutamic acid residues in the vitamin K–dependent factors, a critical step for calcium and phospholipid binding of these proteins (Fig. 141-2). The enzymes γ-glutamylcarboxylase and epoxide reductase are critical for the metabolism and regeneration of vitamin K. Mutations in the genes encoding the γ-carboxylase (GGCX) or vitamin K epoxide reductase complex 1 (VKORC1) result in defective enzymes and thus in vitamin K–dependent factors with reduced activity, varying from 1 to 30% of normal. The disease phenotype is characterized by mild to severe bleeding episodes present from birth. Some patients respond to high doses of vitamin K. For severe bleeding, replacement therapy with FFP or PCC may be necessary to achieve full hemostatic control.
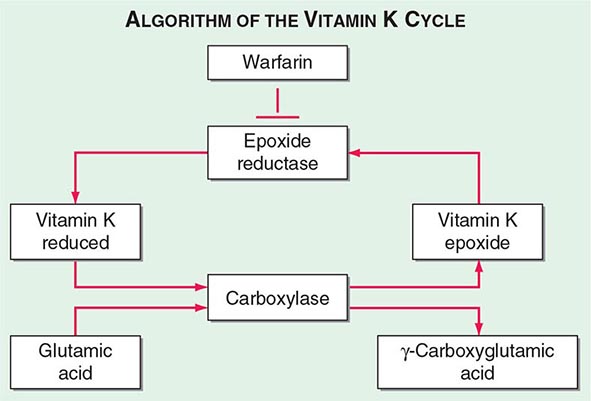
FIGURE 141-2 The vitamin K cycle. Vitamin K is a cofactor for the formation of γ-carboxyglutamic acid residues on coagulation proteins. Vitamin K–dependent γ-glutamylcarboxylase, the enzyme that catalyzes the vitamin K epoxide reductase, regenerates reduced vitamin K. Warfarin blocks the action of the reductase and competitively inhibits the effects of vitamin K.
DISSEMINATED INTRAVASCULAR COAGULATION
DIC is a clinicopathologic syndrome characterized by widespread intravascular fibrin formation in response to excessive blood protease activity that overcomes the natural anticoagulant mechanisms. There are several underlying pathologies associated with DIC (Table 141-2).
COMMON CLINICAL CAUSES OF DISSEMINATED INTRAVASCULAR COAGULATION |
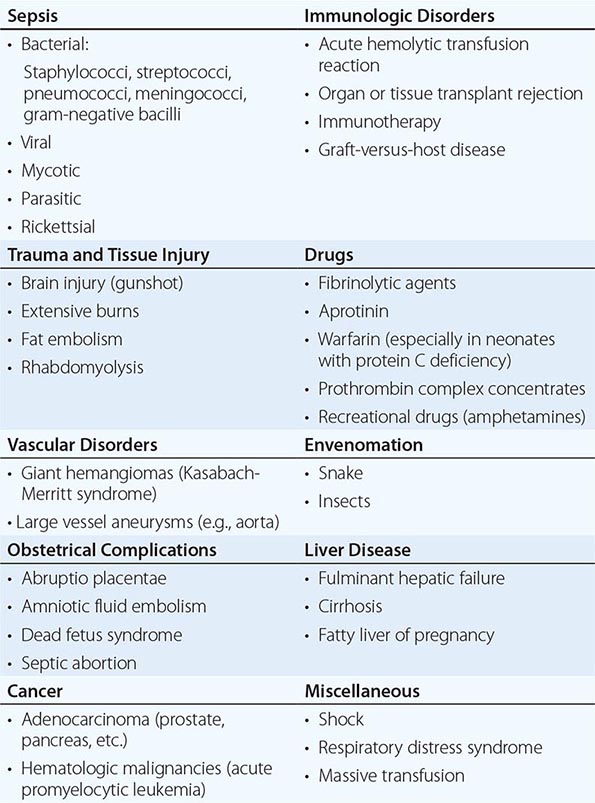
The most common causes are bacterial sepsis, malignant disorders such as solid tumors or acute promyelocytic leukemia, and obstetric causes. DIC is diagnosed in almost one-half of pregnant women with abruptio placentae or with amniotic fluid embolism. Trauma, particularly to the brain, can also result in DIC. The exposure of blood to phospholipids from damaged tissue, hemolysis, and endothelial damage are all contributing factors to the development of DIC in this setting. Purpura fulminans is a severe form of DIC resulting from thrombosis of extensive areas of the skin; it affects predominantly young children following viral or bacterial infection, particularly those with inherited or acquired hypercoagulability due to deficiencies of the components of the protein C pathway. Neonates homozygous for protein C deficiency also present high risk for purpura fulminans with or without thrombosis of large vessels.
The central mechanism of DIC is the uncontrolled generation of thrombin by exposure of the blood to pathologic levels of tissue factor (Fig. 141-3). Simultaneous suppression of physiologic anticoagulant mechanisms and abnormal fibrinolysis further accelerate the process. Together, these abnormalities contribute to systemic fibrin deposition in small and midsize vessels. The duration and intensity of the fibrin deposition can compromise the blood supply of many organs, especially the lung, kidney, liver, and brain, with consequent organ failure. The sustained activation of coagulation results in consumption of clotting factors and platelets, which in turn leads to systemic bleeding. This is further aggravated by secondary hyperfibrinolysis. Studies in animals demonstrate that the fibrinolytic system is indeed suppressed at the time of maximal activation of coagulation. Interestingly, in patients with acute promyelocytic leukemia, a severe hyperfibrinolytic state often occurs in addition to the coagulation activation. The release of several proinflammatory cytokines such as interleukin 6 and tumor necrosis factor α plays a central role in mediating the coagulation defects in DIC and symptoms associated with systemic inflammatory response syndrome (SIRS).
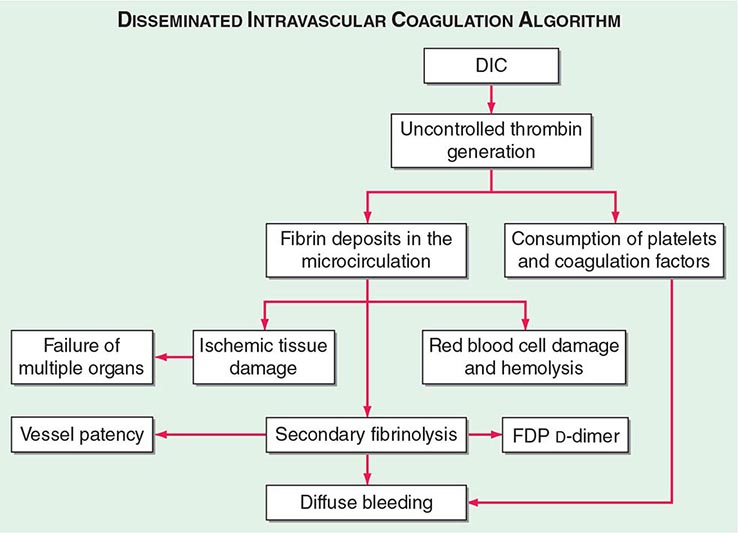
FIGURE 141-3 The pathophysiology of disseminated intravascular coagulation (DIC). Interactions between coagulation and fibrinolytic pathways result in bleeding and thrombosis in the microcirculation in patients with DIC. FDP, fibrin degradation product.
Clinical manifestations of DIC are related to the magnitude of the imbalance of hemostasis, to the underlying disease, or to both. The most common findings are bleeding ranging from oozing from venipuncture sites, petechiae, and ecchymoses to severe hemorrhage from the gastrointestinal tract, lung, or into the CNS. In chronic DIC, the bleeding symptoms are discrete and restricted to skin or mucosal surfaces. The hypercoagulability of DIC manifests as the occlusion of vessels in the microcirculation and resulting organ failure. Thrombosis of large vessels and cerebral embolism can also occur. Hemodynamic complications and shock are common among patients with acute DIC. The mortality ranges from 30 to >80% depending on the underlying disease, the severity of the DIC, and the age of the patient.
The diagnosis of clinically significant DIC is based on the presence of clinical and/or laboratory abnormalities of coagulation or thrombocytopenia. The laboratory diagnosis of DIC should prompt a search for the underlying disease if it is not already apparent. There is no single test that establishes the diagnosis of DIC. The laboratory investigation should include coagulation tests (aPTT, PT, thrombin time [TT]) and markers of fibrin degradation products (FDPs), in addition to platelet and red cell count and analysis of the blood smear. These tests should be repeated over a period of 6–8 h because an initially mild abnormality can change dramatically in patients with severe DIC.
Common findings include the prolongation of PT and/or aPTT; platelet counts μ100,000/μL, or a rapid decline in platelet numbers; the presence of schistocytes (fragmented red cells) in the blood smear; and elevated levels of FDP. The most sensitive test for DIC is the FDP level. DIC is an unlikely diagnosis in the presence of normal levels of FDP. The D-dimer test is more specific for detection of fibrin—but not fibrinogen—degradation products and indicates that the cross-linked fibrin has been digested by plasmin. Because fibrinogen has a prolonged half-life, plasma levels diminish acutely only in severe cases of DIC. High-grade DIC is also associated with levels of antithrombin III or plasminogen activity <60% of normal.
Chronic DIC Low-grade, compensated DIC can occur in clinical situations including giant hemangioma, metastatic carcinoma, or the dead fetus syndrome. Plasma levels of FDP or D-dimers are elevated. aPTT, PT, and fibrinogen values are within the normal range or high. Mild thrombocytopenia or normal platelet counts are also common findings. Red cell fragmentation is often detected but at a lower degree than in acute DIC.
Differential Diagnosis The differential diagnosis between DIC and severe liver disease is challenging and requires serial measurements of the laboratory parameters of DIC. Patients with severe liver disease are at risk for bleeding and manifest laboratory features including thrombocytopenia (due to platelet sequestration, portal hypertension, or hypersplenism), decreased synthesis of coagulation factors and natural anticoagulants, and elevated levels of FDP due to reduced hepatic clearance. However, in contrast to DIC, these laboratory parameters in liver disease do not change rapidly. Other important differential findings include the presence of portal hypertension or other clinical or laboratory evidence of an underlying liver disease.
Microangiopathic disorders such as thrombotic thrombocytopenic purpura present an acute clinical onset of illness accompanied by thrombocytopenia, red cell fragmentation, and multiorgan failure. However, there is no consumption of clotting factors or hyperfibrinolysis.
Over the last few years, several clinical trials on immune therapies for neoplasias using monoclonal antibodies or gene-modified T cells targeting tumor-specific antigens showed unwanted inflammatory responses with increased cytokine release. These complications are sometimes associated with increased D-dimers and decreased fibrinogen levels, cytopenias, and liver dysfunction; thus, careful screening tests for DIC are indicated.



