419 | Diabetes Mellitus: Complications |
Diabetes-related complications affect many organ systems and are responsible for the majority of morbidity and mortality associated with the disease. Strikingly, in the United States, diabetes is the leading cause of new blindness in adults, renal failure, and nontraumatic lower extremity amputation. Diabetes-related complications usually do not appear until the second decade of hyperglycemia. Because type 2 diabetes mellitus (DM) often has a long asymptomatic period of hyperglycemia before diagnosis, many individuals with type 2 DM have complications at the time of diagnosis. Fortunately, many of the diabetes-related complications can be prevented or delayed with early detection, aggressive glycemic control, and efforts to minimize the risks of complications.
Diabetes-related complications can be divided into vascular and nonvascular complications and are similar for type 1 and type 2 DM (Table 419-1). The vascular complications of DM are further subdivided into microvascular (retinopathy, neuropathy, nephropathy) and macrovascular complications (coronary heart disease [CHD], peripheral arterial disease [PAD], cerebrovascular disease). Microvascular complications are diabetes-specific, whereas macrovascular complications are similar to those in nondiabetics but occur at greater frequency in individuals with diabetes. Nonvascular complications include gastroparesis, infections, skin changes, and hearing loss. Whether type 2 DM increases the risk of dementia or impaired cognitive function is not clear.
DIABETES-RELATED COMPLICATIONS |
aThickened skin and reduced joint mobility.
GLYCEMIC CONTROL AND COMPLICATIONS
The microvascular complications of both type 1 and type 2 DM result from chronic hyperglycemia (Fig. 419-1). Evidence implicating a causative role for chronic hyperglycemia in the development of macrovascular complications is less conclusive. CHD events and mortality rate are two to four times greater in patients with type 2 DM and correlate with fasting and postprandial plasma glucose levels as well the hemoglobin A1c (HbA1c). Other factors such as dyslipidemia and hypertension also play important roles in macrovascular complications.
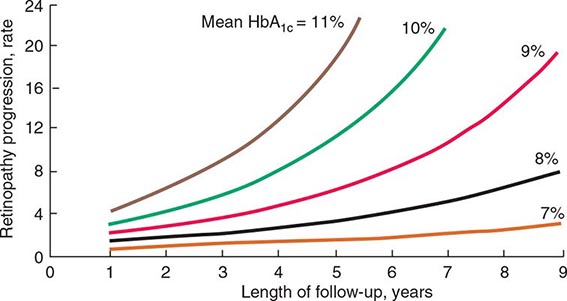
FIGURE 419-1 Relationship of glycemic control and diabetes duration to diabetic retinopathy. The progression of retinopathy in individuals in the Diabetes Control and Complications Trial is graphed as a function of the length of follow-up with different curves for different hemoglobin A1c (HbA1c) values. (Adapted from The Diabetes Control and Complications Trial Research Group: Diabetes 44:968, 1995.)
The Diabetes Control and Complications Trial (DCCT) provided definitive proof that reduction in chronic hyperglycemia can prevent many complications of type 1 DM (Fig. 419-1). This large multicenter clinical trial randomized more than 1400 individuals with type 1 DM to either intensive or conventional diabetes management and prospectively evaluated the development of diabetes-related complications during a mean follow-up of 6.5 years. Individuals in the intensive diabetes management group received multiple administrations of insulin each day (injection or pump) along with extensive educational, psychological, and medical support. Individuals in the conventional diabetes management group received twice-daily insulin injections and quarterly nutritional, educational, and clinical evaluation. The goal in the former group was normoglycemia; the goal in the latter group was prevention of symptoms of diabetes. Individuals in the intensive diabetes management group achieved a substantially lower HbA1c (7.3%) than individuals in the conventional diabetes management group (9.1%). After the DCCT results were reported in 1993, study participants continue to be followed in the Epidemiology of Diabetes Intervention and Complications (EDIC) trial, which recently completed 30 years of follow-up (DCCT + EDIC). At the end of the DCCT phase, study participants in both intensive and conventional arms were offered intensive therapy. However, during the subsequent follow-up of more than 18 years, the initial separation in glycemic control disappeared with both arms maintaining a mean HbA1c of 8.0%.
The DCCT phase demonstrated that improvement of glycemic control reduced nonproliferative and proliferative retinopathy (47% reduction), microalbuminuria (39% reduction), clinical nephropathy (54% reduction), and neuropathy (60% reduction). Improved glycemic control also slowed the progression of early diabetic complications. During the DCCT phase, weight gain (4.6 kg) and severe hypoglycemia (requiring assistance of another person to treat) were more common in the intensive therapy group. The benefits of an improvement in glycemic control occurred over the entire range of HbA1c values (Fig. 419-1), indicating that at any HbA1c level, an improvement in glycemic control is beneficial. The results of the DCCT predicted that individuals in the intensive diabetes management group would gain 7.7 additional years of vision, 5.8 additional years free from end-stage renal disease (ESRD), and 5.6 years free from lower extremity amputations. If all complications of DM were combined, individuals in the intensive diabetes management group would experience 15.3 more years of life without significant microvascular or neurologic complications of DM, compared to individuals who received standard therapy. This translates into an additional 5.1 years of life expectancy for individuals in the intensive diabetes management group. The 30-year follow-up data in the intensively treated group show a continued reduction in retinopathy, nephropathy, and cardiovascular disease. For example, individuals in the intensive therapy group had a 42–57% reduction in cardiovascular events (nonfatal myocardial infarction [MI], stroke, or death from a cardiovascular event) at a mean follow-up of 17 years, even though their subsequent glycemic control was the same as those in the conventional diabetes management group from years 6.5–17. During the EDIC phase, less than 1% of the cohort had become blind, lost a limb to amputation, or required dialysis.
The United Kingdom Prospective Diabetes Study (UKPDS) studied the course of >5000 individuals with type 2 DM for >10 years. This study used multiple treatment regimens and monitored the effect of intensive glycemic control and risk factor treatment on the development of diabetic complications. Newly diagnosed individuals with type 2 DM were randomized to (1) intensive management using various combinations of insulin, a sulfonylurea, or metformin or (2) conventional therapy using dietary modification and pharmacotherapy with the goal of symptom prevention. In addition, individuals were randomly assigned to different antihypertensive regimens. Individuals in the intensive treatment arm achieved an HbA1c of 7%, compared to a 7.9% HbA1c in the standard treatment group. The UKPDS demonstrated that each percentage point reduction in HbA1c was associated with a 35% reduction in microvascular complications. As in the DCCT, there was a continuous relationship between glycemic control and development of complications. Improved glycemic control also reduced the cardiovascular event rate in the follow-up period of >10 years.
One of the major findings of the UKPDS was that strict blood pressure control significantly reduced both macro- and microvascular complications. In fact, the beneficial effects of blood pressure control were greater than the beneficial effects of glycemic control. Lowering blood pressure to moderate goals (144/82 mmHg) reduced the risk of DM-related death, stroke, microvascular endpoints, retinopathy, and heart failure (risk reductions between 32 and 56%).
Similar reductions in the risks of retinopathy and nephropathy were also seen in a small trial of lean Japanese individuals with type 2 DM randomized to either intensive glycemic control or standard therapy with insulin (Kumamoto study). These results demonstrate the effectiveness of improved glycemic control in individuals of different ethnicity and, presumably, a different etiology of DM (i.e., phenotypically different from those in the DCCT and UKPDS). The Action to Control Cardiovascular Risk in Diabetes (ACCORD) and Action in Diabetes and Vascular Disease: Preterax and Diamicron MR Controlled Evaluation (ADVANCE) trials also found that improved glycemic control reduced microvascular complications.
Thus, these large clinical trials in type 1 and type 2 DM indicate that chronic hyperglycemia plays a causative role in the pathogenesis of diabetic microvascular complications. In both the DCCT and the UKPDS, cardiovascular events were reduced at follow-up of >10 years, even though the improved glycemic control was not maintained. The positive impact of a period of improved glycemic control on later disease has been termed a legacy effect or metabolic memory.
A summary of the features of diabetes-related complications includes the following. (1) Duration and degree of hyperglycemia correlate with complications. (2) Intensive glycemic control is beneficial in all forms of DM. (3) Blood pressure control is critical, especially in type 2 DM. (4) Survival in patients with type 1 DM is improving, and diabetes-related complications are declining. (5) Not all individuals with diabetes develop diabetes-related complications. Other incompletely defined factors appear to modulate the development of complications. For example, despite long-standing DM, some individuals never develop nephropathy or retinopathy. Many of these patients have glycemic control that is indistinguishable from those who develop microvascular complications, suggesting a genetic susceptibility for developing particular complications.
MECHANISMS OF COMPLICATIONS
Although chronic hyperglycemia is an important etiologic factor leading to complications of DM, the mechanism(s) by which it leads to such diverse cellular and organ dysfunction is unknown. An emerging hypothesis is that hyperglycemia leads to epigenetic changes (Chap. 82) that influence gene expression in affected cells. For example, this may explain the legacy effect or metabolic memory mentioned above.
Four theories, which are not mutually exclusive, on how hyperglycemia might lead to the chronic complications of DM include the following pathways. (1) Increased intracellular glucose leads to the formation of advanced glycosylation end products, which bind to a cell surface receptor, via the nonenzymatic glycosylation of intra- and extracellular proteins, leading to cross-linking of proteins, accelerated atherosclerosis, glomerular dysfunction, endothelial dysfunction, and altered extracellular matrix composition. (2) Hyperglycemia increases glucose metabolism via the sorbitol pathway related to the enzyme aldose reductase. However, testing of this theory in humans, using aldose reductase inhibitors, has not demonstrated beneficial effects. (3) Hyperglycemia increases the formation of diacylglycerol, leading to activation of protein kinase C, which alters the transcription of genes for fibronectin, type IV collagen, contractile proteins, and extracellular matrix proteins in endothelial cells and neurons. (4) Hyperglycemia increases the flux through the hexosamine pathway, which generates fructose-6-phosphate, a substrate for O-linked glycosylation and proteoglycan production, leading to altered function by glycosylation of proteins such as endothelial nitric oxide synthase or by changes in gene expression of transforming growth factor β (TGF-β) or plasminogen activator inhibitor-1.
Growth factors may play an important role in some diabetes-related complications, and their production is increased by most of these proposed pathways. Vascular endothelial growth factor A (VEGF-A) is increased locally in diabetic proliferative retinopathy and decreases after laser photocoagulation. TGF-β is increased in diabetic nephropathy and stimulates basement membrane production of collagen and fibronectin by mesangial cells. A possible unifying mechanism is that hyperglycemia leads to increased production of reactive oxygen species or superoxide in the mitochondria; these compounds may activate all four of the pathways described above. Although hyperglycemia serves as the initial trigger for complications of diabetes, it is still unknown whether the same pathophysiologic processes are operative in all complications or whether some pathways predominate in certain organs.
OPHTHALMOLOGIC COMPLICATIONS OF DIABETES MELLITUS
DM is the leading cause of blindness between the ages of 20 and 74 in the United States. The gravity of this problem is highlighted by the finding that individuals with DM are 25 times more likely to become legally blind than individuals without DM. Severe vision loss is primarily the result of progressive diabetic retinopathy and clinically significant macular edema. Diabetic retinopathy is classified into two stages: nonproliferative and proliferative. Nonproliferative diabetic retinopathy usually appears late in the first decade or early in the second decade of the disease and is marked by retinal vascular microaneurysms, blot hemorrhages, and cotton-wool spots (Fig. 419-2). Mild nonproliferative retinopathy may progress to more extensive disease, characterized by changes in venous vessel caliber, intraretinal microvascular abnormalities, and more numerous microaneurysms and hemorrhages. The pathophysiologic mechanisms invoked in nonproliferative retinopathy include loss of retinal pericytes, increased retinal vascular permeability, alterations in retinal blood flow, and abnormal retinal microvasculature, all of which can lead to retinal ischemia. A new concept is that the pathology involves inflammatory processes in the retinal neurovascular unit, which consists of neurons, glia, astrocytes, Muüller cells, and specialized vasculature.
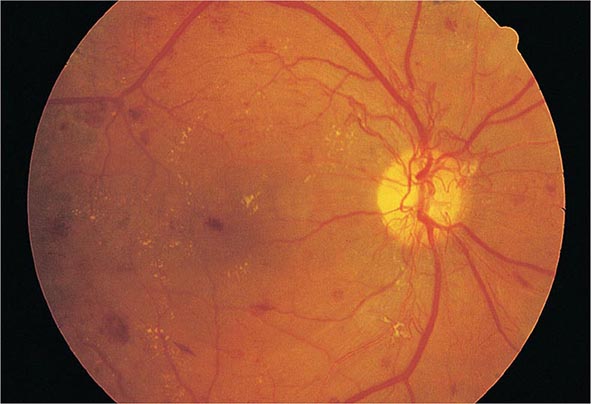
FIGURE 419-2 Diabetic retinopathy results in scattered hemorrhages, yellow exudates, and neovascularization. This patient has neovascular vessels proliferating from the optic disc, requiring urgent panretinal laser photocoagulation.
The appearance of neovascularization in response to retinal hypoxemia is the hallmark of proliferative diabetic retinopathy (Fig. 419-2). These newly formed vessels appear near the optic nerve and/or macula and rupture easily, leading to vitreous hemorrhage, fibrosis, and ultimately retinal detachment. Not all individuals with nonproliferative retinopathy go on to develop proliferative retinopathy, but the more severe the nonproliferative disease, the greater the chance of evolution to proliferative retinopathy within 5 years. This creates an important opportunity for early detection and treatment of diabetic retinopathy. Clinically significant macular edema can occur in the context of nonproliferative or proliferative retinopathy. Fluorescein angiography and optical coherence tomography are useful to detect macular edema, which is associated with a 25% chance of moderate visual loss over the next 3 years. Duration of DM and degree of glycemic control are the best predictors of the development of retinopathy; hypertension and nephropathy are also risk factors. Nonproliferative retinopathy is found in many individuals who have had DM for >20 years. Although there is genetic susceptibility for retinopathy, it confers less influence than either the duration of DM or the degree of glycemic control.
TREATMENT | DIABETIC RETINOPATHY |
The most effective therapy for diabetic retinopathy is prevention. Intensive glycemic and blood pressure control will delay the development or slow the progression of retinopathy in individuals with either type 1 or type 2 DM. Paradoxically, during the first 6–12 months of improved glycemic control, established diabetic retinopathy may transiently worsen. Fortunately, this progression is temporary, and in the long term, improved glycemic control is associated with less diabetic retinopathy. Individuals with known retinopathy may be candidates for prophylactic laser photocoagulation when initiating intensive therapy. Once advanced retinopathy is present, improved glycemic control imparts less benefit, although adequate ophthalmologic care can prevent most blindness.
Regular, comprehensive eye examinations are essential for all individuals with DM (see Table 418-1). Most diabetic eye disease can be successfully treated if detected early. Routine, nondilated eye examinations by the primary care provider or diabetes specialist are inadequate to detect diabetic eye disease, which requires an ophthalmologist for optimal care of these disorders. Laser photocoagulation is very successful in preserving vision. Proliferative retinopathy is usually treated with panretinal laser photocoagulation, whereas macular edema is treated with focal laser photocoagulation and anti–vascular endothelial growth factor therapy (ocular injection). Aspirin therapy (650 mg/d) does not appear to influence the natural history of diabetic retinopathy.
RENAL COMPLICATIONS OF DIABETES MELLITUS
Diabetic nephropathy is the leading cause of chronic kidney disease (CKD), ESRD, and CKD requiring renal replacement therapy. Furthermore, the prognosis of diabetic patients on dialysis is poor, with survival comparable to many forms of cancer. Albuminuria in individuals with DM is associated with an increased risk of cardiovascular disease. Individuals with diabetic nephropathy commonly have diabetic retinopathy.
Like other microvascular complications, the pathogenesis of diabetic nephropathy is related to chronic hyperglycemia. The mechanisms by which chronic hyperglycemia leads to diabetic nephropathy, although incompletely defined, involve the effects of soluble factors (growth factors, angiotensin II, endothelin, advanced glycation end products [AGEs]), hemodynamic alterations in the renal microcirculation (glomerular hyperfiltration or hyperperfusion, increased glomerular capillary pressure), and structural changes in the glomerulus (increased extracellular matrix, basement membrane thickening, mesangial expansion, fibrosis). Some of these effects may be mediated through angiotensin II receptors. Smoking accelerates the decline in renal function. Because only 20–40% of patients with diabetes develop diabetic nephropathy, additional genetic or environmental susceptibility factors remain unidentified. Known risk factors include race and a family history of diabetic nephropathy. Diabetic nephropathy and ESRD secondary to DM develop more commonly in African Americans, Native Americans, and Hispanic individuals with diabetes.
The natural history of diabetic nephropathy is characterized by a fairly predictable sequence of events that was initially defined for individuals with type 1 DM but appears to be similar in type 2 DM (Fig. 419-3). Glomerular hyperperfusion and renal hypertrophy occur in the first years after the onset of DM and are associated with an increase of the glomerular filtration rate (GFR). During the first 5 years of DM, thickening of the glomerular basement membrane, glomerular hypertrophy, and mesangial volume expansion occur as the GFR returns to normal. After 5–10 years of type 1 DM, many individuals begin to excrete small amounts of albumin in the urine. The American Diabetes Association (ADA) recently suggested that the terms previously used to refer to increased urinary protein (microalbuminuria as defined as 30–299 mg/d in a 24-h collection or 30–299 μg/mg creatinine in a spot collection or macroalbuminuria as defined as >300 mg/24 h) be replaced by the phrases “persistent albuminuria (30–299 mg/24 h)” and “persistent albuminuria (≥300 mg/24 h)” to better reflect the continuous nature of albumin excretion in the urine as risk factor for nephropathy and cardiovascular disease (CVD). This chapter uses the terms microalbuminuria and macroalbuminuria. Although the appearance of microalbuminuria in type 1 DM is an important risk factor for progression to macroalbuminuria, only ~50% of individuals progress to macroalbuminuria over the next 10 years. In some individuals with type 1 diabetes and microalbuminuria of short duration, the microalbuminuria regresses. Microalbuminuria is also a risk factor for CVD. Once macroalbuminuria is present, there is a steady decline in GFR, and ~50% of individuals reach ESRD in 7–10 years. Once macroalbuminuria develops, blood pressure rises slightly and the pathologic changes are likely irreversible.
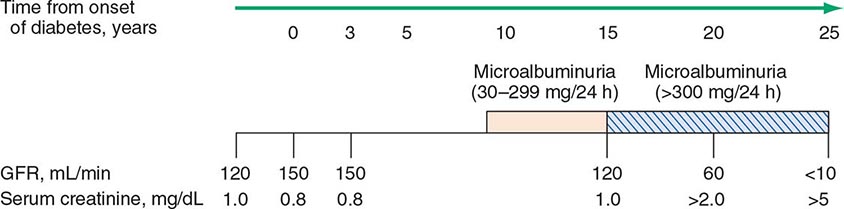
FIGURE 419-3 Time course of development of diabetic nephropathy. The relationship of time from onset of diabetes, the glomerular filtration rate (GFR), and the serum creatinine are shown. (Adapted from RA DeFranzo, in Therapy for Diabetes Mellitus and Related Disorders, 3rd ed. American Diabetes Association, Alexandria, VA, 1998.)
The nephropathy that develops in type 2 DM differs from that of type 1 DM in the following respects: (1) microalbuminuria or macroalbuminuria may be present when type 2 DM is diagnosed, reflecting its long asymptomatic period; (2) hypertension more commonly accompanies microalbuminuria or macroalbuminuria in type 2 DM; and (3) microalbuminuria may be less predictive of diabetic nephropathy and likelihood of progression to macroalbuminuria in type 2 DM, in large part due to increased CV mortality in this population. Finally, it should be noted that albuminuria in type 2 DM may be secondary to factors unrelated to DM, such as hypertension, congestive heart failure (CHF), prostate disease, or infection.
As part of comprehensive diabetes care (Chap. 418), albuminuria should be detected at an early stage when effective therapies can be instituted. Because some individuals with type 1 or type 2 DM have a decline in GFR in the absence of albuminuria, annual measurement of the serum creatinine to estimate GFR should also be performed. An annual microalbuminuria measurement (albumin-to-creatinine ratio in spot urine) is advised in individuals with type 1 or type 2 DM (Fig. 419-4). The urine protein measurement in a routine urinalysis does not detect these low levels of albumin excretion. Screening for albuminuria should commence 5 years after the onset of type 1 DM and at the time of diagnosis of type 2 DM.
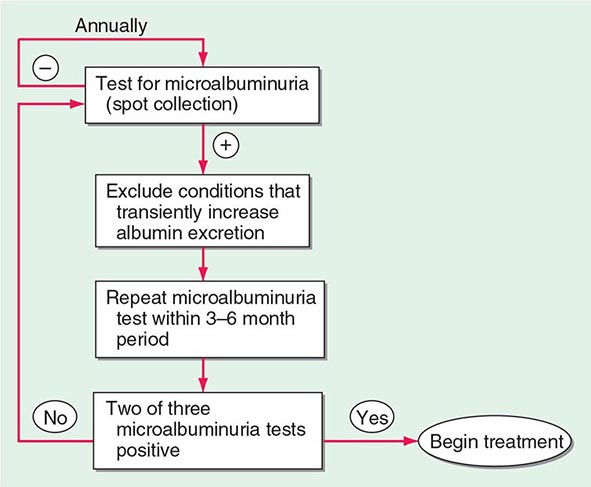
FIGURE 419-4 Screening for microalbuminuria should be performed in patients with type 1 diabetes for ≥5 years, in patients with type 2 diabetes, and during pregnancy. Non-diabetes-related conditions that might increase microalbuminuria are urinary tract infection, hematuria, heart failure, febrile illness, severe hyperglycemia, severe hypertension, and vigorous exercise. (Adapted from RA DeFranzo, in Therapy for Diabetes Mellitus and Related Disorders, 3rd ed. American Diabetes Association, Alexandria, VA, 1998.)
Type IV renal tubular acidosis (hyporeninemic hypoaldosteronism) may occur in type 1 or 2 DM. These individuals develop a propensity to hyperkalemia and acidemia, which may be exacerbated by medications (especially angiotensin-converting enzyme [ACE] inhibitors, angiotensin receptor blockers [ARBs], and spironolactone). Patients with DM are predisposed to radiocontrast-induced nephrotoxicity. Risk factors for radiocontrast-induced nephrotoxicity are preexisting nephropathy and volume depletion. Individuals with DM undergoing radiographic procedures with contrast dye should be well hydrated before and after dye exposure, and the serum creatinine should be monitored for 24–48 h following the procedure. Metformin should be held if indicated.
TREATMENT | DIABETIC NEPHROPATHY |
The optimal therapy for diabetic nephropathy is prevention by control of glycemia (Chap. 418 outlines glycemic goals and approaches). Interventions effective in slowing progression of albuminuria include (1) improved glycemic control, (2) strict blood pressure control, and (3) administration of an ACE inhibitor or ARB. Dyslipidemia should also be treated.
Improved glycemic control reduces the rate at which microalbuminuria appears and progresses in type 1 and type 2 DM. However, once macroalbuminuria is present, it is unclear whether improved glycemic control will slow progression of renal disease. During the later phase of declining renal function, insulin requirements may fall as the kidney is a site of insulin degradation. As the GFR decreases with progressive nephropathy, the use and dose of glucose-lowering agents should be reevaluated (see Table 418-5). Some glucose-lowering medications (sulfonylureas and metformin) are contraindicated in advanced renal insufficiency.
Many individuals with type 1 or type 2 DM develop hypertension. Numerous studies in both type 1 and type 2 DM demonstrate the effectiveness of strict blood pressure control in reducing albumin excretion and slowing the decline in renal function. Blood pressure should be maintained at <140/90 mmHg in diabetic individuals.
Either ACE inhibitors or ARBs should be used to reduce the albuminuria and the associated decline in GFR that accompanies it in individuals with type 1 or type 2 DM (see “Hypertension,” below). Although direct comparisons of ACE inhibitors and ARBs are lacking, most experts believe that the two classes of drugs are equivalent in patient with diabetes. ARBs can be used as an alternative in patients who develop ACE inhibitor–associated cough or angioedema. After 2–3 months of therapy in patients with microalbuminuria, the drug dose is increased until the maximum tolerated dose is reached. Recent studies do not show benefit of intervention prior to onset of microalbuminuria. The combination of an ACE inhibitor and an ARB is not recommended and appears to be detrimental. If use of either ACE inhibitors or ARBs is not possible or the blood pressure is not controlled, then, diuretics, calcium channel blockers (nondihydropyridine class), or beta blockers should be used. These salutary effects are mediated by reducing intraglomerular pressure and inhibition of angiotensin-driven sclerosing pathways, in part through inhibition of TGF-β-mediated pathways.
The ADA does not suggest restriction of protein intake in diabetic individuals with albuminuria because studies have failed to show benefit.
Nephrology consultation should be considered when albuminuria appears and again when the estimated GFR is <60 mL/min per 1.743 m2. As compared with nondiabetic individuals, hemodialysis in patients with DM is associated with more frequent complications, such as hypotension (due to autonomic neuropathy or loss of reflex tachycardia), more difficult vascular access, and accelerated progression of retinopathy. Complications of atherosclerosis are the leading cause of death in diabetic individuals with nephropathy and hyperlipidemia should be treated aggressively. Renal transplantation from a living related donor is the preferred therapy but requires chronic immunosuppression. Combined pancreas-kidney transplant offers the promise of normoglycemia and freedom from dialysis.
NEUROPATHY AND DIABETES MELLITUS
Diabetic neuropathy occurs in ~50% of individuals with long-standing type 1 and type 2 DM. It may manifest as polyneuropathy, mononeuropathy, and/or autonomic neuropathy. As with other complications of DM, the development of neuropathy correlates with the duration of diabetes and glycemic control. Additional risk factors are body mass index (BMI) (the greater the BMI, the greater the risk of neuropathy) and smoking. The presence of CVD, elevated triglycerides, and hypertension is also associated with diabetic peripheral neuropathy. Both myelinated and unmyelinated nerve fibers are lost. Because the clinical features of diabetic neuropathy are similar to those of other neuropathies, the diagnosis of diabetic neuropathy should be made only after other possible etiologies are excluded (Chap. 459).
Polyneuropathy/Mononeuropathy The most common form of diabetic neuropathy is distal symmetric polyneuropathy. It most frequently presents with distal sensory loss and pain, but up to 50% of patients do not have symptoms of neuropathy. Hyperesthesia, paresthesia, and dysesthesia also may occur. Any combination of these symptoms may develop as neuropathy progresses. Symptoms may include a sensation of numbness, tingling, sharpness, or burning that begins in the feet and spreads proximally. Neuropathic pain develops in some of these individuals, occasionally preceded by improvement in their glycemic control. Pain typically involves the lower extremities, is usually present at rest, and worsens at night. Both an acute (lasting <12 months) and a chronic form of painful diabetic neuropathy have been described. The acute form is sometimes treatment-related, occurring in the context of improved glycemic control. As diabetic neuropathy progresses, the pain subsides and eventually disappears, but a sensory deficit in the lower extremities persists. Physical examination reveals sensory loss, loss of ankle deep-tendon reflexes, and abnormal position sense.
Diabetic polyradiculopathy is a syndrome characterized by severe disabling pain in the distribution of one or more nerve roots. It may be accompanied by motor weakness. Intercostal or truncal radiculopathy causes pain over the thorax or abdomen. Involvement of the lumbar plexus or femoral nerve may cause severe pain in the thigh or hip and may be associated with muscle weakness in the hip flexors or extensors (diabetic amyotrophy). Fortunately, diabetic polyradiculopathies are usually self-limited and resolve over 6–12 months.
Mononeuropathy (dysfunction of isolated cranial or peripheral nerves) is less common than polyneuropathy in DM and presents with pain and motor weakness in the distribution of a single nerve. Mononeuropathies can occur at entrapment sites such as carpal tunnel or be noncompressive. A vascular etiology for noncompressive mononeuropathies has been suggested, but the pathogenesis is unknown. Involvement of the third cranial nerve is most common and is heralded by diplopia. Physical examination reveals ptosis and ophthalmoplegia with normal pupillary constriction to light. Sometimes other cranial nerves, such as IV, VI, or VII (Bell’s palsy), are affected. Peripheral mononeuropathies or simultaneous involvement of more than one nerve (mononeuropathy multiplex) may also occur.
Autonomic Neuropathy Individuals with long-standing type 1 or 2 DM may develop signs of autonomic dysfunction involving the cholinergic, noradrenergic, and peptidergic (peptides such as pancreatic polypeptide, substance P, etc.) systems. DM-related autonomic neuropathy can involve multiple systems, including the cardiovascular, gastrointestinal, genitourinary, sudomotor, and metabolic systems. Autonomic neuropathies affecting the cardiovascular system cause a resting tachycardia and orthostatic hypotension. Reports of sudden death have also been attributed to autonomic neuropathy. Gastroparesis and bladder-emptying abnormalities are often caused by the autonomic neuropathy seen in DM (discussed below). Hyperhidrosis of the upper extremities and anhidrosis of the lower extremities result from sympathetic nervous system dysfunction. Anhidrosis of the feet can promote dry skin with cracking, which increases the risk of foot ulcers. Autonomic neuropathy may reduce counterregulatory hormone release (especially catecholamines), leading to an inability to sense hypoglycemia appropriately (hypoglycemia unawareness; Chap. 420), thereby subjecting the patient to the risk of severe hypoglycemia and complicating efforts to improve glycemic control.
TREATMENT | DIABETIC NEUROPATHY |
Treatment of diabetic neuropathy is less than satisfactory. Improved glycemic control should be aggressively pursued and will improve nerve conduction velocity, but symptoms of diabetic neuropathy may not necessarily improve. Efforts to improve glycemic control in long-standing diabetes may be confounded by autonomic neuropathy and hypoglycemia unawareness. Risk factors for neuropathy such as hypertension and hypertriglyceridemia should be treated. Avoidance of neurotoxins (alcohol) and smoking, supplementation with vitamins for possible deficiencies (B12, folate; Chap. 96e), and symptomatic treatment are the mainstays of therapy. Loss of sensation in the foot places the patient at risk for ulceration and its sequelae; consequently, prevention of such problems is of paramount importance. Patients with symptoms or signs of neuropathy should check their feet daily and take precautions (footwear) aimed at preventing calluses or ulcerations. If foot deformities are present, a podiatrist should be involved.
Chronic, painful diabetic neuropathy is difficult to treat but may respond to duloxetine, amitriptyline, gabapentin, valproate, pregabalin, or opioids. Two agents, duloxetine and pregabalin, have been approved by the U.S. Food and Drug Administration (FDA) for pain associated with diabetic neuropathy, but no treatments are satisfactory. No direct comparisons of agents are available, and it is reasonable to switch agents if there is no response or if side effects develop. Referral to a pain management center may be necessary. Because the pain of acute diabetic neuropathy may resolve over time, medications may be discontinued as progressive neuronal damage from DM occurs.
Therapy of orthostatic hypotension secondary to autonomic neuropathy is also challenging. A variety of agents have limited success (fludrocortisone, midodrine, clonidine, octreotide, and yohimbine), but each has significant side effects. Nonpharmacologic maneuvers (adequate salt intake, avoidance of dehydration and diuretics, and lower extremity support hose) may offer some benefit.
GASTROINTESTINAL/GENITOURINARY DYSFUNCTION
Long-standing type 1 and 2 DM may affect the motility and function of the gastrointestinal (GI) and genitourinary systems. The most prominent GI symptoms are delayed gastric emptying (gastroparesis) and altered small- and large-bowel motility (constipation or diarrhea). Gastroparesis may present with symptoms of anorexia, nausea, vomiting, early satiety, and abdominal bloating. Microvascular complications (retinopathy and neuropathy) are usually present. Nuclear medicine scintigraphy after ingestion of a radiolabeled meal may document delayed gastric emptying, but may not correlate well with the patient’s symptoms. Noninvasive “breath tests” following ingestion of a radiolabeled meal have been developed, but are not yet validated. Although parasympathetic dysfunction secondary to chronic hyperglycemia is important in the development of gastroparesis, hyperglycemia itself also impairs gastric emptying. Nocturnal diarrhea, alternating with constipation, is a feature of DM-related GI autonomic neuropathy. In type 1 DM, these symptoms should also prompt evaluation for celiac sprue because of its increased frequency. Esophageal dysfunction in long-standing DM may occur but is usually asymptomatic.
Diabetic autonomic neuropathy may lead to genitourinary dysfunction including cystopathy and female sexual dysfunction (reduced sexual desire, dyspareunia, reduced vaginal lubrication). Symptoms of diabetic cystopathy begin with an inability to sense a full bladder and a failure to void completely. As bladder contractility worsens, bladder capacity and the postvoid residual increase, leading to symptoms of urinary hesitancy, decreased voiding frequency, incontinence, and recurrent urinary tract infections. Diagnostic evaluation includes cystometry and urodynamic studies.
Erectile dysfunction and retrograde ejaculation are very common in DM and may be one of the earliest signs of diabetic neuropathy (Chap. 67). Erectile dysfunction, which increases in frequency with the age of the patient and the duration of diabetes, may occur in the absence of other signs of diabetic autonomic neuropathy.
TREATMENT | GASTROINTESTINAL/GENITOURINARY DYSFUNCTION |
Current treatments for these complications of DM are inadequate. Improved glycemic control should be a primary goal, because some aspects (neuropathy, gastric function) may improve. Smaller, more frequent meals that are easier to digest (liquid) and low in fat and fiber may minimize symptoms of gastroparesis. Metoclopramide has been used but is now restricted in both the United States and Europe and not advised for long-term use. Gastric electrical stimulatory devices are available but not approved. Diabetic diarrhea in the absence of bacterial overgrowth is treated symptomatically (Chap. 349).
Diabetic cystopathy should be treated with scheduled voiding or self-catheterization. Drugs that inhibit type 5 phosphodiesterase are effective for erectile dysfunction, but their efficacy in individuals with DM is slightly lower than in the nondiabetic population (Chap. 67). Sexual dysfunction in women may be improved with use of vaginal lubricants, treatment of vaginal infections, and systemic or local estrogen replacement.
CARDIOVASCULAR MORBIDITY AND MORTALITY
CVD is increased in individuals with type 1 or type 2 DM. The Framingham Heart Study revealed a marked increase in PAD, coronary artery disease, MI, and CHF (risk increase from one- to fivefold) in DM. In addition, the prognosis for individuals with diabetes who have coronary artery disease or MI is worse than for nondiabetics. CHD is more likely to involve multiple vessels in individuals with DM. In addition to CHD, cerebrovascular disease is increased in individuals with DM (threefold increase in stroke). Thus, after controlling for all known cardiovascular risk factors, type 2 DM increases the cardiovascular death rate twofold in men and fourfold in women.
The American Heart Association has designated DM as a “CHD risk equivalent,” and type 2 DM patients without a prior MI have a similar risk for coronary artery–related events as nondiabetic individuals who have had a prior MI. However, the cardiovascular risk assessment in type 2 DM should encompass a more nuanced approach. Cardiovascular risk is lower and not equivalent in a younger individual with a brief duration of type 2 DM compared to an older individual with long-standing type 2DM. Because of the extremely high prevalence of underlying CVD in individuals with diabetes (especially in type 2 DM), evidence of atherosclerotic vascular disease (e.g., cardiac stress test) should be sought in an individual with diabetes who has symptoms suggestive of cardiac ischemia or peripheral or carotid arterial disease. The screening of asymptomatic individuals with diabetes for CHD, even with a risk-factor scale, is not recommended because recent studies have not shown a clinical benefit. The absence of chest pain (“silent ischemia”) is common in individuals with diabetes, and a thorough cardiac evaluation should be considered prior to major surgical procedures.
The increase in cardiovascular morbidity and mortality rates in diabetes appears to relate to the synergism of hyperglycemia with other cardiovascular risk factors. Risk factors for macrovascular disease in diabetic individuals include dyslipidemia, hypertension, obesity, reduced physical activity, and cigarette smoking. Additional risk factors more prevalent in the diabetic population include microalbuminuria, macroalbuminuria, an elevation of serum creatinine, abnormal platelet function and endothelial dysfunction The possibility of atherogenic potential of insulin is suggested by the data in nondiabetic individuals showing higher serum insulin levels (indicative of insulin resistance) in association with greater risk of cardiovascular morbidity and mortality. However, treatment with insulin and the sulfonylureas did not increase the risk of CVD in individuals with type 2 DM.
TREATMENT | CARDIOVASCULAR DISEASE |
In general, the treatment of coronary disease is not different in the diabetic individual (Chap. 293). Revascularization procedures for CHD, including percutaneous coronary interventions (PCI) and coronary artery bypass grafting (CABG), may be less efficacious in the diabetic individual. Initial success rates of PCI in diabetic individuals are similar to those in the nondiabetic population, but diabetic patients have higher rates of restenosis and lower long-term patency and survival rates in older studies.
Aggressive cardiovascular risk modification in all individuals with DM and glycemic control should be individualized, as discussed in Chap. 418. In patients with known CHD and type 2 DM, an ACE inhibitor (or ARB), a statin, and acetylsalicylic acid (ASA; aspirin) should be considered. Past trepidation about using beta blockers in individuals who have diabetes should not prevent use of these agents because they clearly benefit diabetic patients after MI. In patients with CHF, thiazolidinediones should not be used (Chap. 418). However, metformin can be used in patients with stable CHF if the renal function is normal.
Antiplatelet therapy reduces cardiovascular events in individuals with DM who have CHD and is recommended. Current recommendations by the ADA include the use of aspirin for primary prevention of coronary events in diabetic individuals with an increased 10-year cardiovascular risk >10% (at least one risk factor such as hypertension, smoking, family history, albuminuria, or dyslipidemia in men >50 years or women >60 years of age). ASA is not recommended for primary prevention in those with a 10-year cardiovascular risk <10%. The aspirin dose is the same as in nondiabetic individuals.
Cardiovascular Risk Factors • DYSLIPIDEMIA Individuals with DM may have several forms of dyslipidemia (Chap. 421). Because of the additive cardiovascular risk of hyperglycemia and hyperlipidemia, lipid abnormalities should be assessed aggressively and treated as part of comprehensive diabetes care (Chap. 418). The most common pattern of dyslipidemia is hypertriglyceridemia and reduced high-density lipoprotein (HDL) cholesterol levels. DM itself does not increase levels of low-density lipoprotein (LDL), but the small dense LDL particles found in type 2 DM are more atherogenic because they are more easily glycated and susceptible to oxidation.
Almost all treatment studies of diabetic dyslipidemia have been performed in individuals with type 2 DM because of the greater frequency of dyslipidemia in this form of diabetes. Interventional studies have shown that the beneficial effects of LDL reduction with statins are similar in the diabetic and nondiabetic populations. Large prospective trials of primary and secondary intervention for CHD have included some individuals with type 2 DM, and subset analyses have consistently found that reductions in LDL reduce cardiovascular events and morbidity in individuals with DM. No prospective studies have addressed similar questions in individuals with type 1 DM. Because the frequency of CVD is low in children and young adults with diabetes, assessment of cardiovascular risk should be incorporated into the guidelines discussed below.
Based on the guidelines provided by the ADA, priorities in the treatment of dyslipidemia are as follows: (1) lower the LDL cholesterol, (2) raise the HDL cholesterol, and (3) decrease the triglycerides. A treatment strategy depends on the pattern of lipoprotein abnormalities. Initial therapy for all forms of dyslipidemia should include dietary changes, as well as the same lifestyle modifications recommended in the nondiabetic population (smoking cessation, blood pressure control, weight loss, increased physical activity). The dietary recommendations for individuals with DM include increased monounsaturated fat and carbohydrates and reduced saturated fats and cholesterol (Chap. 421). According to guidelines of the ADA, the target lipid values in diabetic individuals (age >40 years) without CVD should be as follows: LDL <2.6 mmol/L (100 mg/dL); HDL >1 mmol/L (40 mg/dL) in men and >13 mmol/L (50 mg/dL) in women; and triglycerides <1.7 mmol/L (150 mg/dL). In patients >40 years, the ADA recommends addition of a statin, regardless of the LDL level, in patients with CHD and those without CHD who have CHD risk factors. Recently released guidelines by the American College of Cardiology (ACC) and American Heart Association (AHA) differ slightly and recommend that diabetic individuals aged 40–75 without CHD and a LDL of 70–189 mg/dl receive “moderate” intensity statin therapy (Chap. 291e). Improvement in glycemic control will lower triglycerides and have a modest beneficial effect by raising HDL.
If the patient is known to have CHD, the ADA recommends an LDL goal of <18 mmol/L (70 mg/dL) as an “option” (in keeping with evidence that such a goal is beneficial in nondiabetic individuals with CHD [Chap. 421]). The ACC/AHA guidelines do not advocate a specific LDL for statin therapy. HMG-CoA reductase inhibitors are the agents of choice for lowering LDL. Combination therapy with an HMG-CoA reductase inhibitor and a fibrate or another lipid-lowering agent (ezetimibe, niacin) may be considered but increases the possibility of side effects such as myositis and has not been shown to be beneficial. Nicotinic acid effectively raises HDL and can be used in patients with diabetes, but may worsen glycemic control and increase insulin resistance and has not been shown to provide additional benefit beyond statin therapy alone. Bile acid–binding resins should not be used if hypertriglyceridemia is present. In large clinical trials, statin usage is associated with a mild increase in the risk of developing type 2 DM. This risk is greatest in individuals with other risk factors for type 2 DM (Chap. 417). However, the cardiovascular benefits of statin use outweigh the mildly increased risk of diabetes.
HYPERTENSION Hypertension can accelerate other complications of DM, particularly CVD, nephropathy, and retinopathy. In targeting a goal of blood pressure of <140/80 mmHg, therapy should first emphasize lifestyle modifications such as weight loss, exercise, stress management, and sodium restriction. The BP goal should be individualized. In some younger individuals, the provider may target a blood pressure of <130/80 mmHg. Realizing that more than one agent is usually required to reach the blood pressure goal, the ADA recommends that all patients with diabetes and hypertension be treated with an ACE inhibitor or an ARB. Subsequently, agents that reduce cardiovascular risk (beta blockers, thiazide diuretics, and calcium channel blockers) should be incorporated into the regimen. ACE inhibitors and ARBs are likely equivalent in most patients with diabetes and renal disease. Serum potassium and renal function should be monitored.
Because of the high prevalence of atherosclerotic disease in individuals with type 2 DM, the possibility of renovascular hypertension should be considered when the blood pressure is not readily controlled.
LOWER EXTREMITY COMPLICATIONS
DM is the leading cause of nontraumatic lower extremity amputation in the United States. Foot ulcers and infections are also a major source of morbidity in individuals with DM. The reasons for the increased incidence of these disorders in DM involve the interaction of several pathogenic factors: neuropathy, abnormal foot biomechanics, PAD, and poor wound healing. The peripheral sensory neuropathy interferes with normal protective mechanisms and allows the patient to sustain major or repeated minor trauma to the foot, often without knowledge of the injury. Disordered proprioception causes abnormal weight bearing while walking and subsequent formation of callus or ulceration. Motor and sensory neuropathy lead to abnormal foot muscle mechanics and to structural changes in the foot (hammer toe, claw toe deformity, prominent metatarsal heads, Charcot joint). Autonomic neuropathy results in anhidrosis and altered superficial blood flow in the foot, which promote drying of the skin and fissure formation. PAD and poor wound healing impede resolution of minor breaks in the skin, allowing them to enlarge and to become infected.
Many individuals with type 2 DM develop a foot ulcer (great toe or metatarsophalangeal areas are most common), and a significant subset who develop an ulceration will ultimately undergo amputation (14–24% risk with that ulcer or subsequent ulceration). Risk factors for foot ulcers or amputation include male sex, diabetes for >10 years, peripheral neuropathy, abnormal structure of foot (bony abnormalities, callus, thickened nails), PAD, smoking, history of previous ulcer or amputation, visual impairment, and poor glycemic control. Large calluses are often precursors to or overlie ulcerations.
TREATMENT | LOWER EXTREMITY COMPLICATIONS |
The optimal therapy for foot ulcers and amputations is prevention through identification of high-risk patients, education of the patient, and institution of measures to prevent ulceration. High-risk patients should be identified during the routine, annual foot examination performed on all patients with DM (see “Ongoing Aspects of Comprehensive Diabetes Care” in Chap. 418). If the monofilament test or one of the other tests is abnormal, the patient is diagnosed with loss of protective sensation (LOPS; Chap. 417). Providers should consider screening for asymptomatic PAD in individuals >50 years of age who have diabetes and other risk factors using ankle-brachial index testing in high-risk individuals (Chap. 302). Patient education should emphasize (1) careful selection of footwear, (2) daily inspection of the feet to detect early signs of poor-fitting footwear or minor trauma, (3) daily foot hygiene to keep the skin clean and moist, (4) avoidance of self-treatment of foot abnormalities and high-risk behavior (e.g., walking barefoot), and (5) prompt consultation with a health care provider if an abnormality arises. Patients at high risk for ulceration or amputation may benefit from evaluation by a foot care specialist. Calluses and nail deformities should be treated by a podiatrist. Interventions directed at risk factor modification include orthotic shoes and devices, callus management, nail care, and prophylactic measures to reduce increased skin pressure from abnormal bony architecture. Attention to other risk factors for vascular disease (smoking, dyslipidemia, hypertension) and improved glycemic control are also important.
Despite preventive measures, foot ulceration and infection are common and represent a serious problem. Due to the multifactorial pathogenesis of lower extremity ulcers, management of these lesions is multidisciplinary and often demands expertise in orthopedics, vascular surgery, endocrinology, podiatry, and infectious diseases. The plantar surface of the foot is the most common site of ulceration. Ulcers may be primarily neuropathic (no accompanying infection) or may have surrounding cellulitis or osteomyelitis. Cellulitis without ulceration is also frequent and should be treated with antibiotics that provide broad-spectrum coverage, including anaerobes (see below).
An infected ulcer is a clinical diagnosis, because superficial culture of any ulceration will likely find multiple possible bacterial species. The infection surrounding the foot ulcer is often the result of multiple organisms, with aerobic gram-positive cocci (staphylococci including MRSA, Group A and B streptococci) being most common and with aerobic gram-negative bacilli and/or obligate anaerobes as co-pathogens.
Gas gangrene may develop in the absence of clostridial infection. Cultures taken from the surface of the ulcer are not helpful; a culture from the debrided ulcer base or from purulent drainage or aspiration of the wound is the most helpful. Wound depth should be determined by inspection and probing with a blunt-tipped sterile instrument. Plain radiographs of the foot should be performed to assess the possibility of osteomyelitis in chronic ulcers that have not responded to therapy. Magnetic resonance imaging (MRI) is the most specific modality, with nuclear medicine scans and labeled white cell studies as alternatives. Surgical debridement is often necessary.
Osteomyelitis is best treated by a combination of prolonged antibiotics (IV, then oral) and/or possibly debridement of infected bone. The possible contribution of vascular insufficiency should be considered in all patients. Peripheral arterial bypass procedures are often effective in promoting wound healing and in decreasing the need for amputation of the ischemic limb (Chap. 302).
A consensus statement from the ADA identified six interventions with demonstrated efficacy in diabetic foot wounds: (1) off-loading, (2) debridement, (3) wound dressings, (4) appropriate use of antibiotics, (5) revascularization, and (6) limited amputation. Off-loading is the complete avoidance of weight bearing on the ulcer, which removes the mechanical trauma that retards wound healing. Bed rest and a variety of orthotic devices or contact casting limit weight bearing on wounds or pressure points. Surgical debridement is important and effective, but clear efficacy of other modalities for wound cleaning (enzymes, soaking, whirlpools) is lacking. Dressings such as hydrocolloid dressings promote wound healing by creating a moist environment and protecting the wound. Antiseptic agents should be avoided. Topical antibiotics are of limited value. Referral for physical therapy, orthotic evaluation, and rehabilitation should occur once the infection is controlled.
Mild or non-limb-threatening infections can be treated with oral antibiotics directed predominantly at methicillin-susceptible staphylococci and streptococci (e.g., dicloxacillin, cephalosporin, amoxicillin/clavulanate). However the increasing prevalence of MRSA often requires the use of clindamycin, doxycycline, or trimethoprim-sulfamethoxazole. Trimethoprim-sulfamethoxazole exhibits less reliable coverage of streptococci than the β-lactams, and diabetic patients may develop adverse effects including acute kidney injury and hyperkalemia. Surgical debridement of necrotic tissue, local wound care (avoidance of weight bearing over the ulcer), and close surveillance for progression of infection are crucial. More severe infections require IV antibiotics as well as bed rest and local wound care. Urgent surgical debridement may be required. Optimization of glycemic control should be a goal. IV antibiotics should provide broad-spectrum coverage directed toward Staphylococcus aureus, including MRSA, streptococci, gram-negative aerobes, and anaerobic bacteria. Initial antimicrobial regimens include vancomycin plus a β-lactam/β-lactamase inhibitor or carbapenem or vancomycin plus a combination of quinolone plus metronidazole. Daptomycin, ceftaroline, or linezolid may be substituted for vancomycin. If the infection surrounding the ulcer is not improving with IV antibiotics, reassessment of antibiotic coverage and reconsideration of the need for surgical debridement or revascularization are indicated. With clinical improvement, oral antibiotics and local wound care can be continued on an outpatient basis with close follow-up.
INFECTIONS
Individuals with DM have a greater frequency and severity of infection. The reasons for this include incompletely defined abnormalities in cell-mediated immunity and phagocyte function associated with hyperglycemia, as well as diminished vascularization. Hyperglycemia aids the colonization and growth of a variety of organisms (Candida and other fungal species). Many common infections are more frequent and severe in the diabetic population, whereas several rare infections are seen almost exclusively in the diabetic population. Examples of this latter category include rhinocerebral mucormycosis, emphysematous infections of the gallbladder and urinary tract, and “malignant” or invasive otitis externa. Invasive otitis externa is usually secondary to P. aeruginosa infection in the soft tissue surrounding the external auditory canal, usually begins with pain and discharge, and may rapidly progress to osteomyelitis and meningitis. These infections should be sought, in particular, in patients presenting with severe hyperglycemia (Chap. 418).
Pneumonia, urinary tract infections, and skin and soft tissue infections are all more common in the diabetic population. In general, the organisms that cause pulmonary infections are similar to those found in the nondiabetic population; however, gram-negative organisms, S. aureus, and Mycobacterium tuberculosis are more frequent pathogens. Urinary tract infections (either lower tract or pyelonephritis) are the result of common bacterial agents such as Escherichia coli, although several yeast species (Candida and Torulopsis glabrata) are commonly observed. Complications of urinary tract infections include emphysematous pyelonephritis and emphysematous cystitis. Bacteriuria occurs frequently in individuals with diabetic cystopathy. Susceptibility to furunculosis, superficial candidal infections, and vulvovaginitis are increased. Poor glycemic control is a common denominator in individuals with these infections. Diabetic individuals have an increased rate of colonization of S. aureus in the skinfolds and nares. Diabetic patients also have a greater risk of postoperative wound infections.
DERMATOLOGIC MANIFESTATIONS
The most common skin manifestations of DM are xerosis and pruritus and are usually relieved by skin moisturizers. Protracted wound healing and skin ulcerations are also frequent complications. Diabetic dermopathy, sometimes termed pigmented pretibial papules, or “diabetic skin spots,” begins as an erythematous macule or papule that evolves into an area of circular hyperpigmentation. These lesions result from minor mechanical trauma in the pretibial region and are more common in elderly men with DM. Bullous diseases, such as bullosa diabeticorum (shallow ulcerations or erosions in the pretibial region), are also seen. Necrobiosis lipoidica diabeticorum is an uncommon disorder, accompanying diabetes in predominantly young women. This usually begins in the pretibial region as an erythematous plaque or papules that gradually enlarge, darken, and develop irregular margins, with atrophic centers and central ulceration. They are often painful. Vitiligo occurs at increased frequency in individuals with type 1 DM. Acanthosis nigricans (hyperpigmented velvety plaques seen on the neck, axilla, or extensor surfaces) is sometimes a feature of severe insulin resistance and accompanying diabetes. Generalized or localized granuloma annulare (erythematous plaques on the extremities or trunk) and scleredema (areas of skin thickening on the back or neck at the site of previous superficial infections) are more common in the diabetic population. Lipoatrophy and lipohypertrophy can occur at insulin injection sites but are now unusual with the use of human insulin.
420 | Hypoglycemia |
Hypoglycemia is most commonly caused by drugs used to treat diabetes mellitus or by exposure to other drugs, including alcohol. However, a number of other disorders, including critical organ failure, sepsis and inanition, hormone deficiencies, non-β-cell tumors, insulinoma, and prior gastric surgery, can cause hypoglycemia (Table 420-1). Hypoglycemia is most convincingly documented by Whipple’s triad: (1) symptoms consistent with hypoglycemia, (2) a low plasma glucose concentration measured with a precise method (not a glucose monitor), and (3) relief of symptoms after the plasma glucose level is raised. The lower limit of the fasting plasma glucose concentration is normally ∼70 mg/dL (∼3.9 mmol/L), but lower venous glucose levels occur normally, late after a meal, during pregnancy, and during prolonged fasting (>24 h). Hypoglycemia can cause serious morbidity; if severe and prolonged, it can be fatal. It should be considered in any patient with episodes of confusion, an altered level of consciousness, or a seizure.
CAUSES OF HYPOGLYCEMIA IN ADULTS |
Source: From PE Cryer et al: J Clin Endocrinol Metab 94:709, 2009. ©The Endocrine Society, 2009.
SYSTEMIC GLUCOSE BALANCE AND GLUCOSE COUNTERREGULATION
Glucose is an obligate metabolic fuel for the brain under physiologic conditions. The brain cannot synthesize glucose or store more than a few minutes’ supply as glycogen and therefore requires a continuous supply of glucose from the arterial circulation. As the arterial plasma glucose concentration falls below the physiologic range, blood-to-brain glucose transport becomes insufficient to support brain energy metabolism and function. However, redundant glucose counterregulatory mechanisms normally prevent or rapidly correct hypoglycemia.
Plasma glucose concentrations are normally maintained within a relatively narrow range—roughly 70–110 mg/dL (3.9–6.1 mmol/L) in the fasting state, with transient higher excursions after a meal—despite wide variations in exogenous glucose delivery from meals and in endogenous glucose utilization by, for example, exercising muscle. Between meals and during fasting, plasma glucose levels are maintained by endogenous glucose production, hepatic glycogenolysis, and hepatic (and renal) gluconeogenesis (Fig. 420-1). Although hepatic glycogen stores are usually sufficient to maintain plasma glucose levels for ∼8 h, this period can be shorter if glucose demand is increased by exercise or if glycogen stores are depleted by illness or starvation.
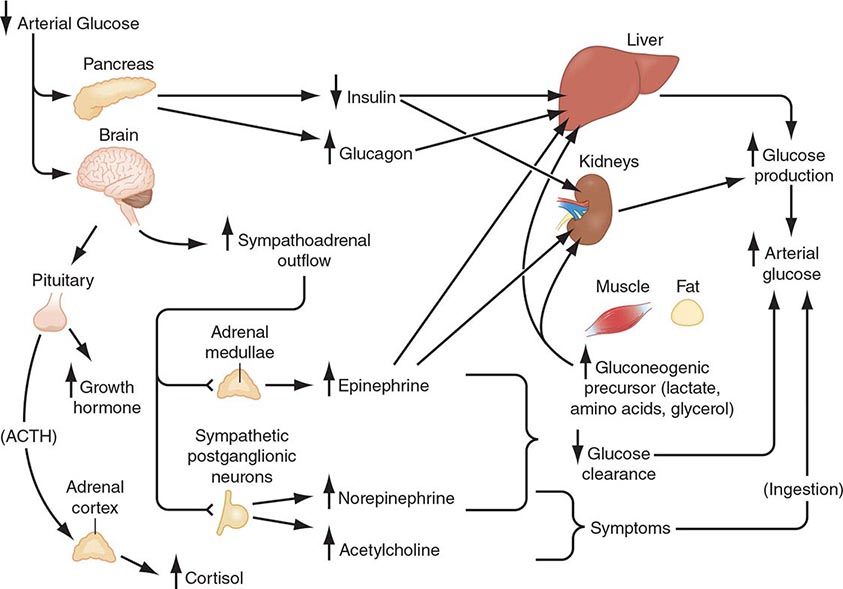
FIGURE 420-1 Physiology of glucose counterregulation: mechanisms that normally prevent or rapidly correct hypoglycemia. In insulin-deficient diabetes, the key counterregulatory responses—suppression of insulin and increases in glucagon—are lost, and stimulation of sympathoadrenal outflow is attenuated. ACTH, adrenocorticotropic hormone.
Gluconeogenesis normally requires low insulin levels and the presence of anti-insulin (counterregulatory) hormones together with a coordinated supply of precursors from muscle and adipose tissue to the liver (and kidneys). Muscle provides lactate, pyruvate, alanine, glutamine, and other amino acids. Triglycerides in adipose tissue are broken down into fatty acids and glycerol, which is a gluconeogenic precursor. Fatty acids provide an alternative oxidative fuel to tissues other than the brain (which requires glucose).
Systemic glucose balance—maintenance of the normal plasma glucose concentration—is accomplished by a network of hormones, neural signals, and substrate effects that regulate endogenous glucose production and glucose utilization by tissues other than the brain (Chap. 417). Among the regulatory factors, insulin plays a dominant role (Table 420-2; Fig. 420-1). As plasma glucose levels decline within the physiologic range in the fasting state, pancreatic β-cell insulin secretion decreases, thereby increasing hepatic glycogenolysis and hepatic (and renal) gluconeogenesis. Low insulin levels also reduce glucose utilization in peripheral tissues, inducing lipolysis and proteolysis and consequently releasing gluconeogenic precursors. Thus, a decrease in insulin secretion is the first defense against hypoglycemia.
PHYSIOLOGIC RESPONSES TO DECREASING PLASMA GLUCOSE CONCENTRATIONS |
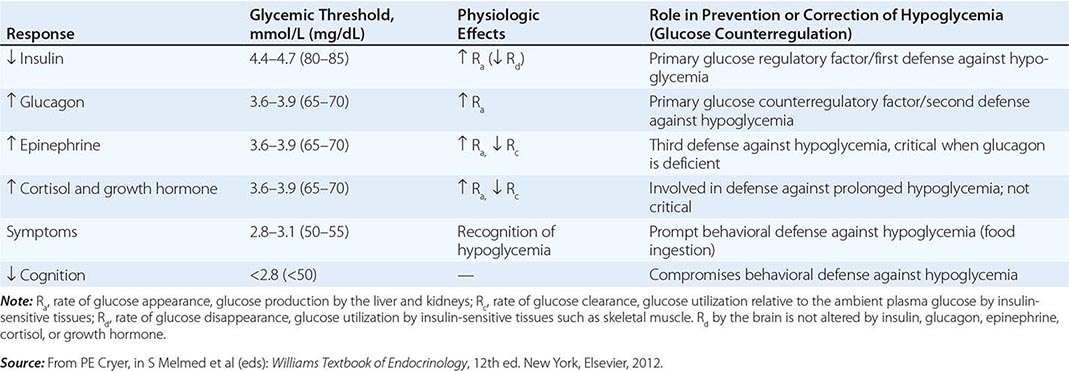
As plasma glucose levels decline just below the physiologic range, glucose counterregulatory (plasma glucose–raising) hormones are released (Table 420-2; Fig. 420-1). Among these, pancreatic α-cell glucagon, which stimulates hepatic glycogenolysis, plays a primary role. Glucagon is the second defense against hypoglycemia. Adrenomedullary epinephrine, which stimulates hepatic glycogenolysis and gluconeogenesis (and renal gluconeogenesis), is not normally critical. However, it becomes critical when glucagon is deficient. Epinephrine is the third defense against hypoglycemia. When hypoglycemia is prolonged beyond ∼4 h, cortisol and growth hormone also support glucose production and restrict glucose utilization to a limited amount (∼20% compared to epinephrine). Thus cortisol and growth hormone play no role in defense against acute hypoglycemia.
As plasma glucose levels fall further, symptoms prompt behavioral defense against hypoglycemia, including the ingestion of food (Table 420-2; Fig. 420-1). The normal glycemic thresholds for these responses to decreasing plasma glucose concentrations are shown in Table 420-2. However, these thresholds are dynamic. They shift to higher-than-normal glucose levels in people with poorly controlled diabetes, who can experience symptoms of hypoglycemia when their glucose levels decline toward the normal range (pseudohypoglycemia). On the other hand, thresholds shift to lower-than-normal glucose levels in people with recurrent hypoglycemia; e.g., patients with aggressively treated diabetes or an insulinoma have symptoms at glucose levels lower than those that cause symptoms in healthy individuals.
Clinical Manifestations Neuroglycopenic manifestations of hypoglycemia are the direct result of central nervous system glucose deprivation. These features include behavioral changes, confusion, fatigue, seizure, loss of consciousness, and, if hypoglycemia is severe and prolonged, death. Neurogenic (or autonomic) manifestations of hypoglycemia result from the perception of physiologic changes caused by the central nervous system–mediated sympathoadrenal discharge that is triggered by hypoglycemia. They include adrenergic symptoms (mediated largely by norepinephrine released from sympathetic postganglionic neurons but perhaps also by epinephrine released from the adrenal medullae), such as palpitations, tremor, and anxiety, as well as cholinergic symptoms (mediated by acetylcholine released from sympathetic postganglionic neurons), such as sweating, hunger, and paresthesias. Clearly, these are nonspecific symptoms. Their attribution to hypoglycemia requires that the corresponding plasma glucose concentration be low and that the symptoms resolve after the glucose level is raised (as delineated by Whipple’s triad).
Common signs of hypoglycemia include diaphoresis and pallor. Heart rate and systolic blood pressure are typically increased but may not be raised in an individual who has experienced repeated, recent episodes of hypoglycemia. Neuroglycopenic manifestations are often observable. Transient focal neurologic deficits occur occasionally. Permanent neurologic deficits are rare.
Etiology and Pathophysiology Hypoglycemia is most commonly a result of the treatment of diabetes. This topic is therefore addressed before other causes of hypoglycemia are considered.
HYPOGLYCEMIA IN DIABETES
Impact and Frequency Hypoglycemia is the limiting factor in the glycemic management of diabetes mellitus. First, it causes recurrent morbidity in most people with type 1 diabetes (T1DM) and in many with advanced type 2 diabetes (T2DM), and it is sometimes fatal. Second, it precludes maintenance of euglycemia over a lifetime of diabetes and thus full realization of the well-established microvascular benefits of glycemic control. Third, it causes a vicious cycle of recurrent hypoglycemia by producing hypoglycemia-associated autonomic failure—i.e., the clinical syndromes of defective glucose counterregulation and of hypoglycemia unawareness (see later).
Hypoglycemia is a fact of life for people with T1DM. They suffer an average of two episodes of symptomatic hypoglycemia per week and at least one episode of severe, at least temporarily disabling hypoglycemia each year. An estimated 6–10% of people with T1DM die as a result of hypoglycemia. The incidence of hypoglycemia is lower in T2DM than in T1DM. However, its prevalence in insulin-requiring T2DM is surprisingly high. Recent studies investigating insulin-pump or multiple-injection therapies have revealed a hypoglycemia prevalence approaching 70%. In fact, as patients with T2DM outnumber those with T1DM by ten- to twentyfold, the prevalence of hypoglycemia is now greater in T2DM. Insulin, a sulfonylurea, or a glinide can cause hypoglycemia in T2DM. Metformin, thiazolidinediones, α-glucosidase inhibitors, glucagon-like peptide 1 (GLP-1) receptor agonists, and dipeptidyl peptidase IV (DPP-IV) inhibitors should not cause hypoglycemia. However, they increase the risk when combined with one of the sulfonylureas or glinides, or with insulin. Notably, the frequency of hypoglycemia approaches that in T1DM as persons with T2DM develop absolute insulin deficiency and require more complex treatment with insulin.
Conventional Risk Factors The conventional risk factors for hypoglycemia in diabetes are identified on the basis of the premise that relative or absolute insulin excess is the sole determinant of risk. Relative or absolute insulin excess occurs when (1) insulin (or insulin secretagogue) doses are excessive, ill-timed, or of the wrong type; (2) the influx of exogenous glucose is reduced (e.g., during an overnight fast or after missed meals or snacks); (3) insulin-independent glucose utilization is increased (e.g., during exercise); (4) sensitivity to insulin is increased (e.g., with improved glycemic control, in the middle of the night, late after exercise, or with increased fitness or weight loss); (5) endogenous glucose production is reduced (e.g., after alcohol ingestion); and (6) insulin clearance is reduced (e.g., in renal failure). However, these conventional risk factors alone explain a minority of episodes; other factors are typically involved.
Hypoglycemia-Associated Autonomic Failure (HAAF) While marked insulin excess alone can cause hypoglycemia, iatrogenic hypoglycemia in diabetes is typically the result of the interplay of relative or absolute therapeutic insulin excess and compromised physiologic and behavioral defenses against falling plasma glucose concentrations (Table 420-2; Fig. 420-2). Defective glucose counterregulation compromises physiologic defense (particularly decrements in insulin and increments in glucagon and epinephrine), and hypoglycemia unawareness compromises behavioral defense (ingestion of carbohydrate).
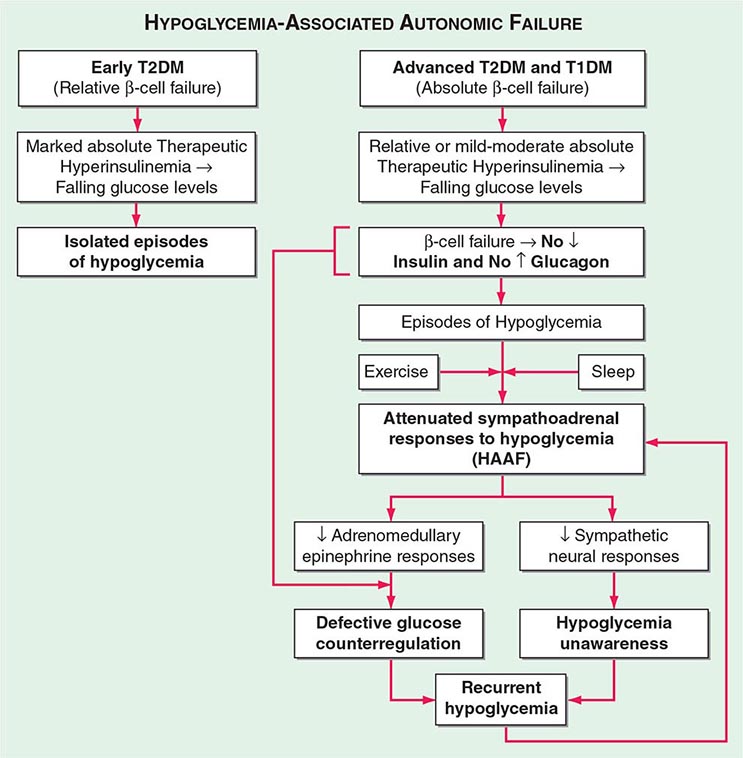
FIGURE 420-2 Hypoglycemia-associated autonomic failure (HAAF) in insulin-deficient diabetes. T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus. (Modified from PE Cryer: Hypoglycemia in Diabetes. Pathophysiology, Prevalence, and Prevention, 2nd ed. © American Diabetes Association, 2012.)
DEFECTIVE GLUCOSE COUNTERREGULATION In the setting of absolute endogenous insulin deficiency, insulin levels do not decrease as plasma glucose levels fall; the first defense against hypoglycemia is lost. Furthermore, probably because the decrement in intraislet insulin is normally a signal to stimulate glucagon secretion, glucagon levels do not increase as plasma glucose levels fall further; a second defense against hypoglycemia is lost. Finally, the increase in epinephrine levels, a third defense against hypoglycemia, in response to a given level of hypoglycemia is typically attenuated. The glycemic threshold for the sympathoadrenal (adrenomedullary epinephrine and sympathetic neural norepinephrine) response is shifted to lower plasma glucose concentrations. That shift is typically the result of recent antecedent iatrogenic hypoglycemia. In the setting of absent decrements in insulin and of absent increments in glucagon, the attenuated increment in epinephrine causes the clinical syndrome of defective glucose counterregulation. Affected patients are at ≥25-fold greater risk of severe iatrogenic hypoglycemia during aggressive glycemic therapy for their diabetes than are patients with normal epinephrine responses. This functional—and potentially reversible—disorder is distinct from classic diabetic autonomic neuropathy—a structural and irreversible disorder.
HYPOGLYCEMIA UNAWARENESS The attenuated sympathoadrenal response (largely the reduced sympathetic neural response) to hypoglycemia causes the clinical syndrome of hypoglycemia unawareness—i.e., loss of the warning adrenergic and cholinergic symptoms that previously allowed the patient to recognize developing hypoglycemia and therefore to abort the episode by ingesting carbohydrates. Affected patients are at a sixfold increased risk of severe iatrogenic hypoglycemia during aggressive glycemic therapy of their diabetes.
HAAF IN DIABETES The concept of HAAF in diabetes posits that recent antecedent iatrogenic hypoglycemia (or sleep or prior exercise) causes both defective glucose counterregulation (by reducing the epinephrine response to a given level of subsequent hypoglycemia in the setting of absent insulin and glucagon responses) and hypoglycemia unawareness (by reducing the sympathoadrenal response to a given level of subsequent hypoglycemia). These impaired responses create a vicious cycle of recurrent iatrogenic hypoglycemia (Fig. 420-2). Hypoglycemia unawareness and, to some extent, the reduced epinephrine component of defective glucose counterregulation are reversible by as little as 2–3 weeks of scrupulous avoidance of hypoglycemia in most affected patients.
On the basis of this pathophysiology, additional risk factors for hypoglycemia in diabetes include (1) absolute insulin deficiency, indicating that insulin levels will not decrease and glucagon levels will not increase as plasma glucose levels fall; (2) a history of severe hypoglycemia or of hypoglycemia unawareness, implying recent antecedent hypoglycemia, as well as prior exercise or sleep, indicating that the sympathoadrenal response will be attenuated; and (3) lower hemoglobin A1c (HbA1c) levels or lower glycemic goals that, all other factors being equal, increase the probability of recent antecedent hypoglycemia.
Hypoglycemia Risk Factor Reduction Several recent multicenter, randomized, controlled trials investigating the potential benefits of tight glucose control in either inpatient or outpatient settings have reported a high prevalence of severe hypoglycemia. In the NICE-SUGAR study, attempts to control in-hospital plasma glucose values towards physiologic levels resulted in increased mortality risk. The ADVANCE and ACCORD studies and the Veterans Affairs Diabetes Trial (VADT) also found a significant incidence of severe hypoglycemia among T2DM patients. Severe hypoglycemia with accompanying serious cardiovascular morbidity and mortality also occurred in the standard (e.g., not receiving intensified treatment) control group in both the ACCORD study and the VADT. Thus, severe hypoglycemia can and does occur at HbA1c values of 8–9% in both T1DM and T2DM. Somewhat surprisingly, all three studies found little or no benefit of intensive glucose control to reduce macrovascular events in T2DM. In fact, the ACCORD study was ended early because of the increased mortality rate in the intensive glucose control arm. Whether iatrogenic hypoglycemia was the cause of the increased mortality risk is not known. In light of these findings, some new recommendations and paradigms have been formulated. Whereas there is little debate regarding the need to reduce hyperglycemia in the hospital, the glycemic maintenance goals have been modified to lie between 140 and 180 mg/dL. Accordingly, the benefits of insulin therapy and reduced hyperglycemia can be obtained while the prevalence of hypoglycemia is reduced.
Similarly, evidence exists that intensive glucose control can reduce the prevalence of microvascular disease in both T1DM and T2DM. These benefits need to be weighed against the increased prevalence of hypoglycemia. Certainly, the level of glucose control (i.e., the HbA1c level) should be evaluated for each patient. Multicenter trials have demonstrated that individuals with recently diagnosed T1DM or T2DM can have better glycemic control with less hypoglycemia. In addition, there is still long-term benefit in reducing HbA1c values from higher to lower, albeit still above recommended levels. Perhaps a reasonable therapeutic goal is the lowest HbA1c level that does not cause severe hypoglycemia and that preserves awareness of hypoglycemia.
Pancreatic transplantation (both whole-organ and islet-cell) has been used in part as a treatment for severe hypoglycemia. Generally, rates of hypoglycemia are reduced after transplantation. This decrease appears to be due to increased physiologic insulin and glucagon responses during hypoglycemia.
The use of continuous glucose monitors offers some promise as a method of reducing hypoglycemia while improving HbA1c. Other interventions to stimulate counterregulatory responses, such as selective serotonin-reuptake inhibitors, β-adrenergic receptor antagonists, opiate receptor antagonists, and fructose, remain experimental and have not been assessed in large-scale clinical trials.
Thus, intensive glycemic therapy (Chap. 418) needs to be applied along with the patient’s education and empowerment, frequent self-monitoring of blood glucose, flexible insulin (and other drug) regimens (including the use of insulin analogues, both short- and longer-acting), individualized glycemic goals, and ongoing professional guidance, support, and consideration of both the conventional risk factors and those indicative of compromised glucose counterregulation. Given a history of hypoglycemia unawareness, a 2- to 3-week period of scrupulous avoidance of hypoglycemia is indicated.
HYPOGLYCEMIA WITHOUT DIABETES
There are many causes of hypoglycemia (Table 420-1). Because hypoglycemia is common in insulin- or insulin secretagogue–treated diabetes, it is often reasonable to assume that a clinically suspicious episode is the result of hypoglycemia. On the other hand, because hypoglycemia is rare in the absence of relevant drug-treated diabetes, it is reasonable to conclude that a hypoglycemic disorder is present only in patients in whom Whipple’s triad can be demonstrated.
Particularly when patients are ill or medicated, the initial diagnostic focus should be on the possibility of drug involvement and then on critical illnesses, hormone deficiency, or non–islet cell tumor hypoglycemia. In the absence of any of these etiologic factors and in a seemingly well individual, the focus should shift to possible endogenous hyperinsulinism or accidental, surreptitious, or even malicious hypoglycemia.
Drugs Insulin and insulin secretagogues suppress glucose production and stimulate glucose utilization. Ethanol blocks gluconeogenesis but not glycogenolysis. Thus, alcohol-induced hypoglycemia typically occurs after a several-day ethanol binge during which the person eats little food, with consequent glycogen depletion. Ethanol is usually measurable in blood at the time of presentation, but its levels correlate poorly with plasma glucose concentrations. Because gluconeogenesis becomes the predominant route of glucose production during prolonged hypoglycemia, alcohol can contribute to the progression of hypoglycemia in patients with insulin-treated diabetes.
Many other drugs have been associated with hypoglycemia. These include commonly used drugs such as angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists, β-adrenergic receptor antagonists, quinolone antibiotics, indomethacin, quinine, and sulfonamides.
Critical Illness Among hospitalized patients, serious illnesses such as renal, hepatic, or cardiac failure; sepsis; and inanition are second only to drugs as causes of hypoglycemia.
Rapid and extensive hepatic destruction (e.g., toxic hepatitis) causes fasting hypoglycemia because the liver is the major site of endogenous glucose production. The mechanism of hypoglycemia in patients with cardiac failure is unknown. Hepatic congestion and hypoxia may be involved. Although the kidneys are a source of glucose production, hypoglycemia in patients with renal failure is also caused by the reduced clearance of insulin and the reduced mobilization of gluconeogenic precursors in renal failure.
Sepsis is a relatively common cause of hypoglycemia. Increased glucose utilization is induced by cytokine production in macrophage-rich tissues such as the liver, spleen, and lung. Hypoglycemia develops if glucose production fails to keep pace. Cytokine-induced inhibition of gluconeogenesis in the setting of nutritional glycogen depletion, in combination with hepatic and renal hypoperfusion, may also contribute to hypoglycemia.
Hypoglycemia can be seen with starvation, perhaps because of loss of whole-body fat stores and subsequent depletion of gluconeogenic precursors (e.g., amino acids), necessitating increased glucose utilization.
Hormone Deficiencies Neither cortisol nor growth hormone is critical to the prevention of hypoglycemia, at least in adults. Nonetheless, hypoglycemia can occur with prolonged fasting in patients with primary adrenocortical failure (Addison’s disease) or hypopituitarism. Anorexia and weight loss are typical features of chronic cortisol deficiency and likely result in glycogen depletion. Cortisol deficiency is associated with impaired gluconeogenesis and low levels of gluconeogenic precursors; these associations suggest that substrate-limited gluconeogenesis, in the setting of glycogen depletion, is the cause of hypoglycemia. Growth hormone deficiency can cause hypoglycemia in young children. In addition to extended fasting, high rates of glucose utilization (e.g., during exercise or in pregnancy) or low rates of glucose production (e.g., after alcohol ingestion) can precipitate hypoglycemia in adults with previously unrecognized hypopituitarism.
Hypoglycemia is not a feature of the epinephrine-deficient state that results from bilateral adrenalectomy when glucocorticoid replacement is adequate, nor does it occur during pharmacologic adrenergic blockade when other glucoregulatory systems are intact. Combined deficiencies of glucagon and epinephrine play a key role in the pathogenesis of iatrogenic hypoglycemia in people with insulin-deficient diabetes, as discussed earlier. Otherwise, deficiencies of these hormones are not usually considered in the differential diagnosis of a hypoglycemic disorder.
Non-β-Cell Tumors Fasting hypoglycemia, often termed non–islet cell tumor hypoglycemia, occurs occasionally in patients with large mesenchymal or epithelial tumors (e.g., hepatomas, adrenocortical carcinomas, carcinoids). The glucose kinetic patterns resemble those of hyperinsulinism (see next), but insulin secretion is suppressed appropriately during hypoglycemia. In most instances, hypoglycemia is due to overproduction of an incompletely processed form of insulin-like growth factor II (“big IGF-II”) that does not complex normally with circulating binding proteins and thus more readily gains access to target tissues. The tumors are usually apparent clinically, plasma ratios of IGF-II to IGF-I are high, and free IGF-II levels (and levels of pro-IGF-II [1–21]) are elevated. Curative surgery is seldom possible, but reduction of tumor bulk may ameliorate hypoglycemia. Therapy with a glucocorticoid, a growth hormone, or both has also been reported to alleviate hypoglycemia. Hypoglycemia attributed to ectopic IGF-I production has been reported but is rare.
Endogenous Hyperinsulinism Hypoglycemia due to endogenous hyperinsulinism can be caused by (1) a primary β-cell disorder—typically a β-cell tumor (insulinoma), sometimes multiple insulinomas, or a functional β-cell disorder with β-cell hypertrophy or hyperplasia; (2) an antibody to insulin or to the insulin receptor; (3) a β-cell secretagogue such as a sulfonylurea; or perhaps (4) ectopic insulin secretion, among other very rare mechanisms. None of these causes is common.
The fundamental pathophysiologic feature of endogenous hyperinsulinism caused by a primary β-cell disorder or an insulin secretagogue is the failure of insulin secretion to fall to very low levels during hypoglycemia. This feature is assessed by measurement of plasma insulin, C-peptide (the connecting peptide that is cleaved from proinsulin to produce insulin), proinsulin, and glucose concentrations during hypoglycemia. Insulin, C-peptide, and proinsulin levels need not be high relative to normal, euglycemic values; rather, they are inappropriately high in the setting of a low plasma glucose concentration. Critical diagnostic findings are a plasma insulin concentration ≥3 μU/mL (≥18 pmol/L), a plasma C-peptide concentration ≥0.6 ng/mL (≥0.2 nmol/L), and a plasma proinsulin concentration ≥5.0 pmol/L when the plasma glucose concentration is <55 mg/dL (<3.0 mmol/L) with symptoms of hypoglycemia. A low plasma β-hydroxybutyrate concentration (≤2.7 mmol/L) and an increment in plasma glucose level of >25 mg/dL (>1.4 mmol/L) after IV administration of glucagon (1.0 mg) indicate increased insulin (or IGF) actions.
The diagnostic strategy is (1) to measure plasma glucose, insulin, C-peptide, proinsulin, and β-hydroxybutyrate concentrations and to screen for circulating oral hypoglycemic agents during an episode of hypoglycemia and (2) to assess symptoms during the episode and seek their resolution following correction of hypoglycemia by IV injection of glucagon (i.e., to document Whipple’s triad). This is straightforward if the patient is hypoglycemic when seen. Since endogenous hyperinsulinemic disorders usually, but not invariably, cause fasting hypoglycemia, a diagnostic episode may develop after a relatively short outpatient fast. Serial sampling during an inpatient diagnostic fast of up to 72 h or after a mixed meal is more problematic. An alternative is to give patients a detailed list of the required measurements and ask them to present to an emergency room, with the list, during a symptomatic episode. Obviously, a normal plasma glucose concentration during a symptomatic episode indicates that the symptoms are not the result of hypoglycemia.
An insulinoma—an insulin-secreting pancreatic islet β-cell tumor—is the prototypical cause of endogenous hyperinsulinism and therefore should be sought in patients with a compatible clinical syndrome. However, insulinoma is not the only cause of endogenous hyperinsulinism. Some patients with fasting endogenous hyperinsulinemic hypoglycemia have diffuse islet involvement with β-cell hypertrophy and sometimes hyperplasia. This pattern is commonly referred to as nesidioblastosis, although β-cells budding from ducts are not invariably found. Other patients have a similar islet pattern but with postprandial hypoglycemia, a disorder termed noninsulinoma pancreatogenous hypoglycemia. Postgastric bypass postprandial hypoglycemia, which most often follows Roux-en-Y gastric bypass, is also characterized by diffuse islet involvement and endogenous hyperinsulinism. Some have suggested that exaggerated GLP-1 responses to meals cause hyperinsulinemia and hypoglycemia, but the relevant pathogenesis has not been clearly established. If medical treatments with agents such as an α-glucosidase inhibitor, diazoxide, or octreotide fail, partial pancreatectomy may be required. Autoimmune hypoglycemias include those caused by an antibody to insulin that binds post-meal insulin and then gradually disassociates, with consequent late postprandial hypoglycemia. Alternatively, an insulin receptor antibody can function as an agonist. The presence of an insulin secretagogue, such as a sulfonylurea or a glinide, results in a clinical and biochemical pattern similar to that of an insulinoma but can be distinguished by the presence of the circulating secretagogue. Finally, there are reports of very rare phenomena such as ectopic insulin secretion, a gain-of-function insulin receptor mutation, and exercise-induced hyperinsulinemia.
Insulinomas are uncommon, with an estimated yearly incidence of 1 in 250, 000. Because more than 90% of insulinomas are benign, they are a treatable cause of potentially fatal hypoglycemia. The median age at presentation is 50 years in sporadic cases, but the tumor usually presents in the third decade when it is a component of multiple endocrine neoplasia type 1 (Chap. 408). More than 99% of insulinomas are within the substance of the pancreas, and the tumors are usually small (<2.0 cm in diameter in 90% of cases). Therefore, they come to clinical attention because of hypoglycemia rather than mass effects. CT or MRI detects ∼70–80% of insulinomas. These methods detect metastases in the roughly 10% of patients with a malignant insulinoma. Transabdominal ultrasound often identifies insulinomas, and endoscopic ultrasound has a sensitivity of ∼90%. Somatostatin receptor scintigraphy is thought to detect insulinomas in about half of patients. Selective pancreatic arterial calcium injections, with the endpoint of a sharp increase in hepatic venous insulin levels, regionalize insulinomas with high sensitivity, but this invasive procedure is seldom necessary except to confirm endogenous hyperinsulinism in the diffuse islet disorders. Intraoperative pancreatic ultrasonography almost invariably localizes insulinomas that are not readily palpable by the surgeon. Surgical resection of a solitary insulinoma is generally curative. Diazoxide, which inhibits insulin secretion, or the somatostatin analogue octreotide can be used to treat hypoglycemia in patients with unresectable tumors; everolimus, an mTOR (mammalian target of rapamycin) inhibitor, is promising.
ACCIDENTAL, SURREPTITIOUS, OR MALICIOUS HYPOGLYCEMIA
Accidental ingestion of an insulin secretagogue (e.g., as the result of a pharmacy or other medical error) or even accidental administration of insulin can occur. Factitious hypoglycemia, caused by surreptitious or even malicious administration of insulin or an insulin secretagogue, shares many clinical and laboratory features with insulinoma. It is most common among health care workers, patients with diabetes or their relatives, and people with a history of other factitious illnesses. However, it should be considered in all patients being evaluated for hypoglycemia of obscure cause. Ingestion of an insulin secretagogue causes hypoglycemia with increased C-peptide levels, whereas exogenous insulin causes hypoglycemia with low C-peptide levels reflecting suppression of insulin secretion.
Analytical error in the measurement of plasma glucose concentrations is rare. On the other hand, glucose monitors used to guide treatment of diabetes are not quantitative instruments, particularly at low glucose levels, and should not be used for the definitive diagnosis of hypoglycemia. Even with a quantitative method, low measured glucose concentrations can be artifactual—e.g., the result of continued glucose metabolism by the formed elements of the blood ex vivo, particularly in the presence of leukocytosis, erythrocytosis, or thrombocytosis or with delayed separation of the serum from the formed elements (pseudohypoglycemia).
INBORN ERRORS OF METABOLISM CAUSING HYPOGLYCEMIA
Nondiabetic hypoglycemia also results from inborn errors of metabolism. Such hypoglycemia most commonly occurs in infancy but can also occur in adulthood. Cases in adults can be classified into those resulting in fasting hypoglycemia, postprandial hypoglycemia, and exercise-induced hypoglycemia.
Fasting Hypoglycemia Although rare, disorders of glycogenolysis can result in fasting hypoglycemia. These disorders include glycogen storage disease (GSD) of types 0, I, III, and IV and Fanconi-Bickel syndrome (Chap. 433e). Patients with GSD types I and III characteristically have high blood lactate levels before and after meals, respectively. Both groups have hypertriglyceridemia, but ketones are high in GSD type III. Defects in fatty acid oxidation also result in fasting hypoglycemia. These defects can include (1) defects in the carnitine cycle; (2) fatty-acid β-oxidation disorders; (3) electron transfer disturbances; and (4) ketogenesis disorders. Finally, defects in gluconeogenesis (fructose-1, 6-biphosphatase) have been reported to result in recurrent hypoglycemia and lactic acidosis.
Postprandial Hypoglycemia Inborn errors of metabolism resulting in postprandial hypoglycemia are also rare. These errors include (1) glucokinase, SUR1, and Kir6.2 potassium channel mutations; (2) congenital disorders of glycosylation; and (3) inherited fructose intolerance.
Exercise-Induced Hypoglycemia Exercise-induced hypoglycemia, by definition, follows exercise. It results in hyperinsulinemia caused by increased activity of monocarboxylate transporter 1 in β cells.



