CHAPTER 6
Digoxin
JOHN NOVIASKY, PharmD, BCPS
WILLIAM DARKO, BPharm, MPSG, PharmD
DAROWAN AKAJABOR, PharmD, BCPS
ARKADIY MAKARON, PharmD, BCPS
CHRISTOPHER MILLER, PharmD, BCPS, AAHIVP
DEIRDRE P. PIERCE PharmD, BCPS, CGP
ROBERT SEABURY, PharmD
KAREN WHALEN, BS, Pharm, BCPS
Digitalis is the oldest cardiovascular compound still in use today.1 More than 200 years ago, Sir William Withering observed that foxglove flower derivative (digitalis purpura) could be used for “cardiac dropsy.”2 Since that time, the positive hemodynamic, neurohormonal, and electrophysiologic effects of digoxin have been well explored.1
The pharmacodynamic effects of digoxin include increased cardiac output, decreased pulmonary capillary wedge pressure, and increased ejection fraction. The neurohormonal effects of digoxin include improved baroreceptor sensitivity, decreased norepinephrine concentration, decreased renin-angiotensin activations, sympathoinhibitory effect, and increase release of atrial natriuretic peptide and brain natriuretic peptide. The electrophysiological effects are primarily mediated through the interaction of digoxin with the sodium-potassium-ATPase pump.1,2
Digoxin increases contractility by inhibiting the sodium-potassium exchange in the sodium-potassium-ATPase pump leading to an increase of sodium in the myocytes. This increase results in decreased outflow of calcium from the myocyte and greater contractile force of the myocardium.1,2
Digoxin also has electrophysiological effects. It has the ability to slow conduction through the AV node and it can slow the sinus rate via the S-A node. It is these effects that allow it to be used for atrial fibrillation.3
PHARMACOKINETIC PARAMETERS
The bioavailability of digoxin can range from 70 percent to nearly 100 percent, depending on the type of oral formulation. The elixir and tablet formulations are approximately 80 percent and 70 percent bioavailable, respectively.4 Encapsulated digoxin solution is close to 100 percent bioavailable, but no longer manufactured. The bioavailability of intravenous digoxin is always complete. The estimated elimination half-life of digoxin can take as long as 48 hours.5
Digoxin is roughly 30 percent protein bound in the plasma and has a large volume of distribution (VD) of nearly 7 L/kg in healthy adults.5,6 It follows a two-compartment kinetic model with an initial distribution phase into the central compartment consisting primarily of plasma and highly perfused tissues, such as the liver. A second, slower distribution phase soon occurs and moves the drug out of the central compartment and into the peripheral, deep tissue compartment.5 The target site, the myocardium, is affected by drug concentration in the peripheral compartment and, therefore, clinical effect may not be seen until sufficient drug has accumulated at that site, which may take several hours after a loading dose. Serum drug concentrations early after a loading dose may not represent the true drug concentration at the site of action and may lead to inappropriate dosage adjustments.
The VD in obese patients best correlates with ideal body weight, rather than actual body weight.7 Due to digoxin’s hydrophilic nature, it does not significantly distribute into adipose tissue. However, actual body weight should be used in underweight patients, whose ideal body weight is greater than their actual body weight. Pediatric patients have a higher percentage of total body water and, therefore, would have an increased volume of distribution relative to their adult counterparts.
METABOLISM AND ELIMINATION
Digoxin is primarily eliminated via renal excretion as unchanged drug but does undergo hepatic metabolism to a small extent.5,8 Roughly 15–20 percent of the drug is metabolized, with digoxigenin bisdigitoxoside and digoxigenin monodigitoxoside being the primary metabolites. The metabolic pathway for digoxin includes sequential hydrolysis followed by conjugation and oxidation into polar metabolites.5,9 The cytochrome P450 isoenzyme system is responsible for some of digoxin’s metabolism, but only to a minor extent. As such, drug interactions with agents that inhibit or induce CYP450 are minimal. Digoxin’s metabolites have limited cardioactive properties when compared to the parent compound and these metabolites primarily undergo renal elimination.9
Roughly 80 percent of a digoxin dose is eliminated as unchanged drug by the kidneys. Renal elimination of digoxin occurs through a mixture of glomerular filtration and active tubular secretion.5,8-10 Tubular secretion is mediated primarily by the P-glycoprotein transporter and is subject to drug interaction risks. For patients, dosing and safety concerns are highly dependent upon renal function. Patients with severe renal dysfunction may encounter drug accumulation and toxicity. Monitoring of renal function is necessary for patients receiving digoxin therapy, and creatinine clearance estimates remain the most important clinical tool to ensure appropriate dosing. Specific dosing adjustments are recommended for patients with decreased creatinine clearance.11 Due to its high volume of distribution, digoxin is removed negligibly by hemodialysis, and clinical data have indicated higher mortality rates for digoxin-treated patients who require hemodialysis.12,13 Extreme caution, and possibly alternative therapy, is required for these patients.
THERAPEUTIC CONCENTRATIONS
CHRONIC HEART FAILURE (CHF)
Historically, higher concentrations and wider targets were accepted for patients on digoxin, but contemporary literature suggests a more conservative approach in patients with heart failure. The new desired therapeutic range is lower and narrower at 0.5–0.9 ng/mL or less than 1 ng/mL.14 Recent studies suggest that in addition to the positive inotropic effects of digoxin, neurohormonal modulation through inhibition of Na-K ATPase is evident at low digoxin concentrations. Further decreases in norepinephrine concentrations are not observed as the concentration of digoxin increases. In other words, digoxin serum concentrations between 0.7 ng/mL and 1.2 ng/mL attain therapeutic benefit while decreasing risk of toxicity in patients with CHF.15,16
ATRIAL FIBRILLATION (AF)
In patients with atrial fibrillation, the traditionally accepted range of digoxin serum concentration is 0.5–2 ng/mL, but achieving rate control may require targeting higher serum concentrations. In some patients, concentrations >2 ng/mL may be required to adequately control ventricular rate. However, higher levels are more often associated with toxicity (See DIGOXIN TOXICITY Section).
In general, the routine measurement of digoxin serum concentrations is not always necessary. Digoxin toxicity and explaining poor response to therapy are the main indications for measurement of serum digoxin concentrations. Clinical response to therapy should always be considered first.11,17
SAMPLING
Following a loading dose, serum digoxin concentration should be obtained at approximately 6–8 hours following the loading dose. Levels obtained earlier may be falsely elevated due to the slow distribution phase. Once steady state has been achieved, which usually occurs in about 7 to 14 days after a maintenance regimen is initiated or changed, routine samples for digoxin monitoring should be drawn just before the next dose is due.4,11
DOSING
LOADING DOSE
Loading doses are generally unnecessary and not required when digoxin is used to treat CHF. In this patient population, digoxin is used for its positive inotropic effects and neurohormonal modulation. It should be initiated at a maintenance dose dependent on factors such as age, lean body weight, and renal function.16 In atrial fibrillation, digoxin loading dose may be administered at the onset of therapy to achieve a rapid attainment of target concentration. The advantage of giving a loading dose must be weighed against the disadvantage of exposing a patient to an abrupt toxic concentration of digoxin. To achieve adequate response and avoid toxicity, prescribers must account for patient-specific dosing characteristics such as lean body weight, age, renal function, and concomitant medications. These factors will vary from patient to patient.1,3,18
If a loading dose is used, it should be given in divided dosing to decrease the occurrence of toxic concentrations. Intravenous doses can be given in 2- to 4-hour intervals while oral formulations can be given in 6- to 8-hour intervals. For example, a 1 mg digoxin oral loading dose can be given as a 0.5 mg dose followed by a 0.25 mg dose every 6 hours for 2 doses with careful monitoring of the patient for efficacy and toxicity.
A loading dose can be calculated using the following equation:
LD = (VD)(C in ng/mL)/(F)
where VD is the volume of distribution, C is the desired concentration, and F is the bioavailability of the formulation.
Example I
MS is a 47-year-old female to be initiated on intravenous digoxin for atrial fibrillation. Her weight is 60 kg and height is 65 inches. Her CrCl is estimated to be at 95 mL/min. Calculate her digoxin loading dose for a desired plasma concentration of 1 ng/mL.
Loading dose = (VD)(C)/(F)
First calculate VD (using the Jusko equation).
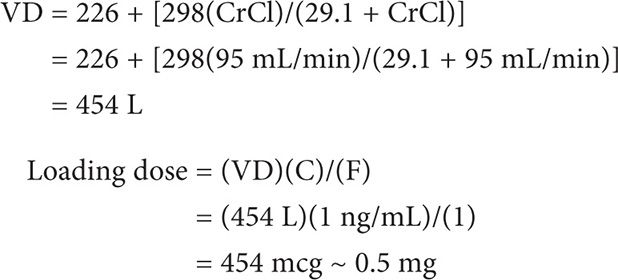
MAINTENANCE DOSE
Most patients with CHF will achieve the target serum digoxin concentration of 0.5 to 1.0 ng/mL with doses of 0.125 to 0.25 mg daily. The dosing nomogram designed by Bauman and colleagues and the dosing formula by Koup and Jusko have been found to be the best methods of estimating digoxin dose in this modern era of new therapeutic range of digoxin in heart failure patients.18,19 The new dosing nomogram for digoxin in patients with heart failure (see Figure 6-1) also takes into account ideal body weight in kilograms, creatinine clearance in mL/min, and height in inches to estimate the dose of digoxin.18
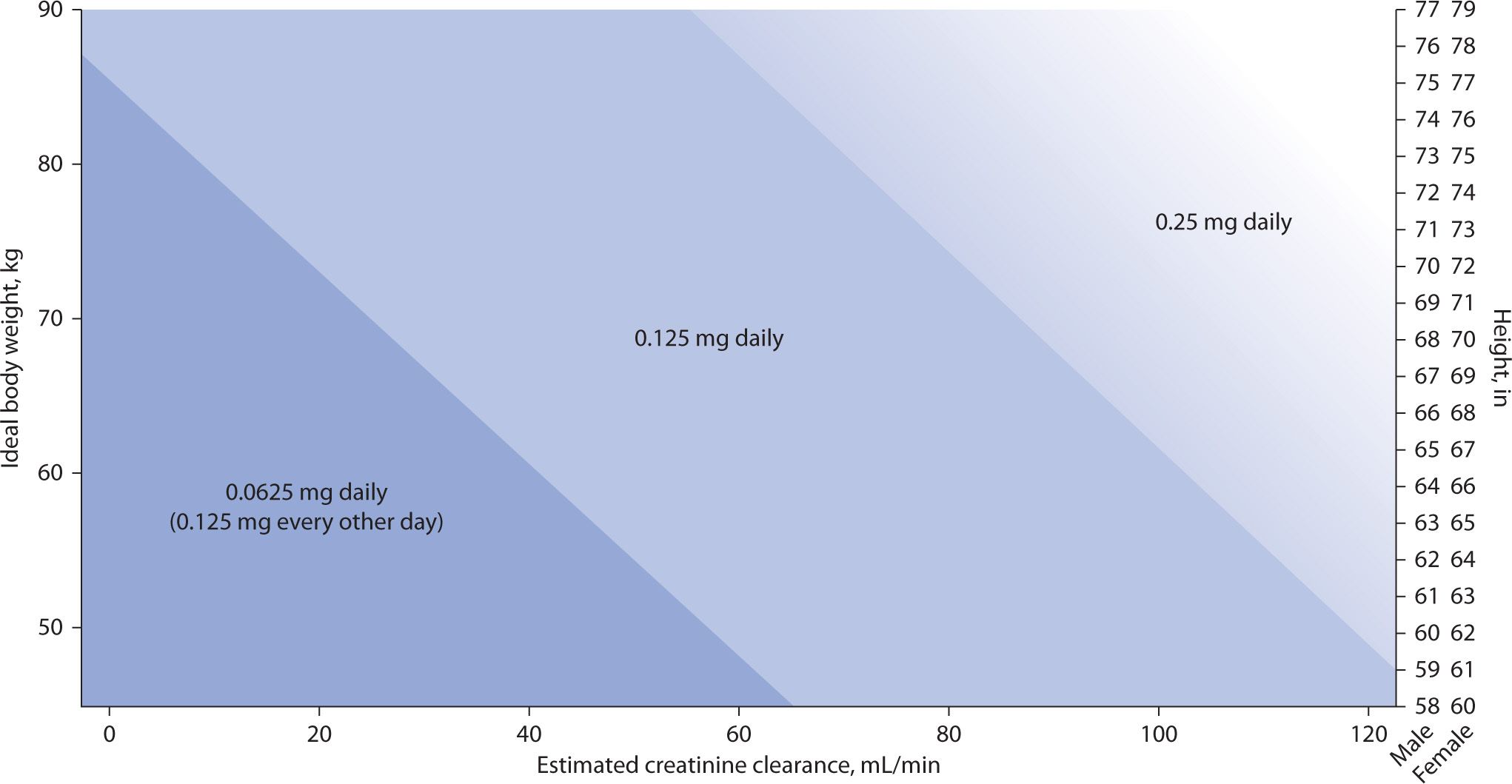
FIGURE 6-1. Dosing nomogram for digoxin in patients with heart failure.18 Note: To select an appropriate digoxin maintenance dose, plot a patient’s creatinine clearance (x-axis) (to convert creatinine clearance to milliliter per second, multiply by 0.01667) and ideal body weight (y-axis). The point at which these lines intersect is the recommended digoxin dose. If ideal body weight has not or cannot be calculated, an appropriate digoxin maintenance dose can be determined by plotting a patient’s creatinine clearance (x-axis) and height (z-axis), depending on patient sex. In the 0.25 mg daily area of the nomogram, one may also consider a digoxin maintenance dose of 0.125 mg, alternating with 0.25 mg every other day (average daily dose of 0.1875 mg/day) as represented by the gradual shading of this area.
The maintenance dose equation takes into account age, gender, and renal function, which are known variables that can alter digoxin concentrations. The maintenance dose can be calculated using the maintenance dose equation: Maintenance dose = (CL)(Css)(T)/(F), where CL is the clearance of digoxin, Css is the steady state concentration, T is the dosing interval in days and F is the bioavailability of the formulation.
Example II
Calculate a maintenance dose (oral tablets) for MS in Example 1.
Maintenance dose = (CL)(Css)(T)/(F)
First calculate CL.

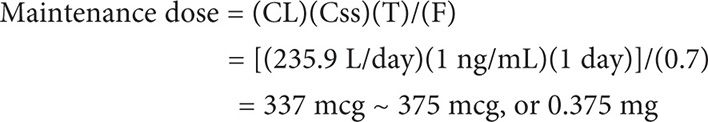
MS can be initiated on 0.25mg (1 tablet) or 0.375 mg (1½ tablets) daily.
MAINTENANCE DOSE BASED ON SERUM DIGOXIN CONCENTRATION
A more patient-specific maintenance dose can be determined using the patient’s dosing history and the observed digoxin concentrations to derive a patient-specific drug clearance.
The equation for maintenance dose, MD = (CL)(Css)(T)/(F), can be rearranged to derive CL.
CL = (MD)(F)/(Css)(T)
Example III
Assuming MS (from Example II) was initiated on 0.375 mg tablets daily and in 14 days a steady-state serum digoxin concentration was obtained right before the next scheduled dose. The Css was measured to be 2.8 ng/mL. Calculate her true CL and a new dosing regimen to attain a new desired Css of 1.5 ng/mL.
First calculate CL.

PHARMACOKINETIC MODIFICATIONS FOR DISEASE STATES
Renal Disease
Due to extensive elimination by the kidney, any reduction in renal function will result in decreased elimination and potential accumulation of this narrow therapeutic index drug. Renal clearance of digoxin has been shown to be similar to creatinine clearance in patients with normal and impaired renal function.20
Patients with renal disease have a lower VD.21 Although this pharmacokinetic change occurs, evidence indicates that myocardial and serum drug concentrations are still an acceptable estimate of digoxin efficacy and toxicity.22 Decreases in VD and elimination by the kidney substantiate the need for lower digoxin loading and maintenance doses, and consideration to dosage interval extension. Because of the large volume of distribution of digoxin, removal by hemodialysis and peritoneal dialysis is minimal and not of benefit as a treatment for toxicity.11,23
Geriatrics
As aging occurs, renal excretion decreases as the number of effective nephrons is reduced and renal blood flow diminishes. Glomerular filtration rate declines 25–50 percent between the ages of 20 and 90.24 In the patient without identified kidney disease, renal elimination of digoxin will still decrease with age. Consequently, the elderly patient should be managed as though they have recognized renal disease.25
Digoxin is largely distributed into skeletal muscle. Older patients experience a 10–20 percent decrease in volume of distribution of digoxin, attributed to loss of lean muscle mass. Additionally, age-related reduction in renal function will contribute to lower VD.24 As a result, lower loading and maintenance doses and careful interval assessment are recommended.
Heart Failure
As compared to healthy adults, the clearance of digoxin is reduced 50 percent in those patients with heart failure as defined by these equations26:

These equations are alternatives for clearance determination first illustrated in example II of Maintenance Dose section.
Bauman and colleagues developed a linear regression model to predict digoxin doses and intervals to target a serum level of 0.7 and subsequently constructed a dosing nomogram using height, ideal body weight, and creatinine clearance.18 (See Figure 6-1.)
Hyperthyroidism
In hyperthyroid disease, myocardial sensitivity and response to digoxin are reduced.20 Additionally, pharmacokinetics can change as VD is increased, which generally supports use of higher loading doses.27 Enhanced elimination of digoxin occurs due to increases in creatinine clearance seen with these patients.28
Hypothyroidism
Conversely, VD of digoxin is reduced in hypothyroidism, suggesting that lower loading doses may be prudent. Clearance is also lower, which is attributed to reduced creatinine clearance evident in that disease state.28
Liver Disease
Adjustment of digoxin dosing is not required in liver disease patients. Pharmacokinetic parameters are not significantly changed.29
DRUG INTERACTIONS
Digoxin has multiple clinically significant drug interactions that alter clearance and result in supratherapeutic or subtherapeutic serum concentrations (Table 6-1). The mechanism of clearance changes is a result of interaction with the P-glycoprotein transporter system.30-31 P-glycoprotein is normally located at the blood-brain barrier, liver, pancreas, placenta, testis, kidney, colon, and jejunum. It is suggested that the purpose of P-glycoprotein is protection against toxic compounds by excretion into urine, bile, and intestinal lumen.30 When this transport system is inhibited or induced, substrates such as digoxin will not be excreted at the same rate, and serum concentrations will increase or decrease respectively.
| TABLE 6-1 | Digoxin Drug Interactions: Summary of Studies |
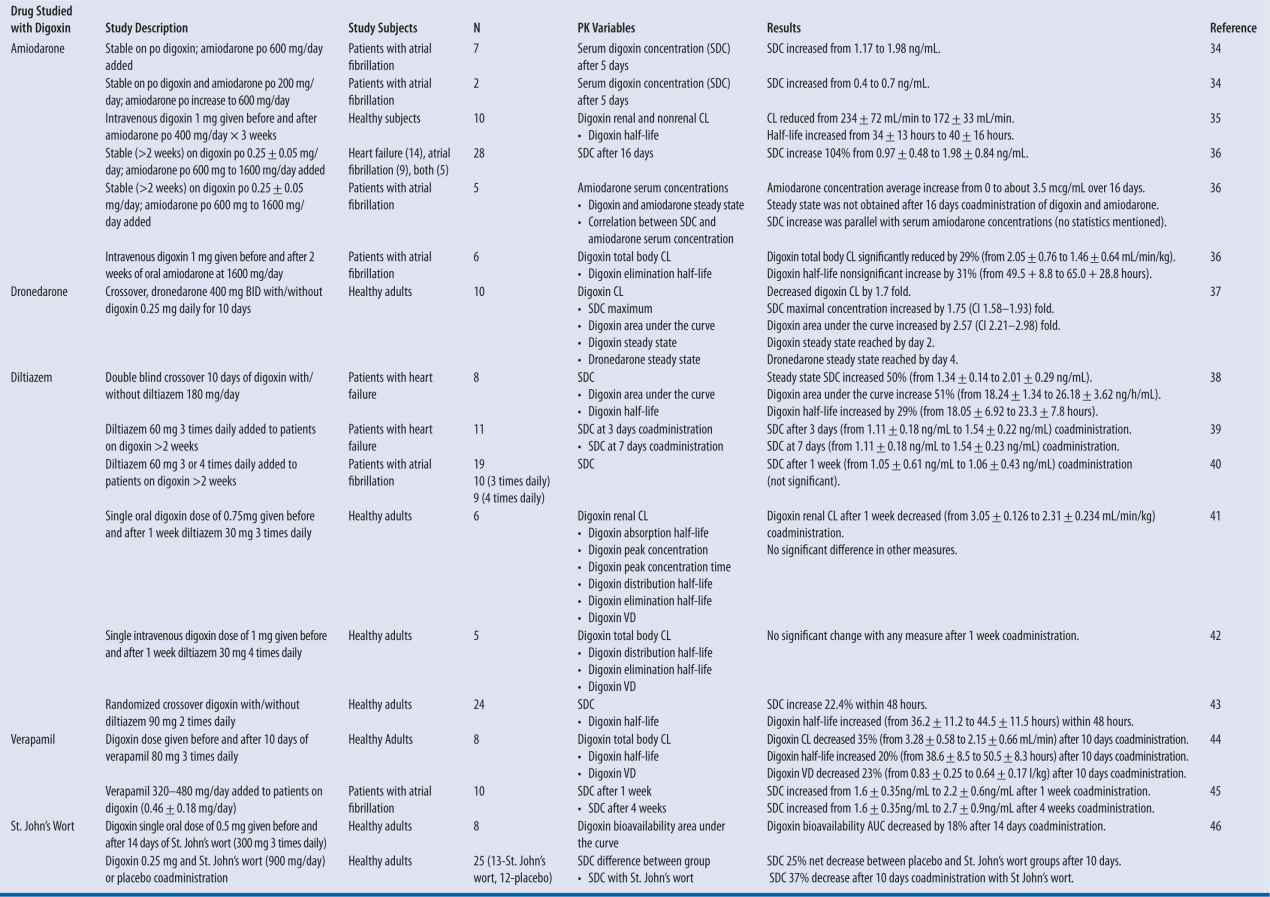
Note: All data shown in results column represents a statistically significant change unless otherwise noted.
Specific P-glycoprotein inhibitors that are expected to increase digoxin exposure by more than 25 percent include amiodarone, captopril, carvedilol, clarithromycin, conivaptan, cyclosporine, diltiazem, dronedarone, felodipine, itraconazole, lopinavir and ritonavir, quinidine, ranolazine, ticagrelor, and verapamil. Conversely, those agents that induce p-glycoprotein enzyme and decrease digoxin area under the curve by greater than 20 percent are phenytoin, rifampin, St. John’s wort, and tipranavir/ritonavir.32-47
A pronounced interaction is seen when quinidine is added to a patient stabilized on digoxin, resulting in decreased volume of distribution and decreased clearance of digoxin. As a result, digoxin serum concentration increases rapidly and in a sustained fashion. Clinically, a reasonable plan is to decrease digoxin dose by 50 percent.33 More commonly, cardiovascular drugs that will be used along with digoxin, and with potential to alter serum concentration, include amiodarone, dronedarone, diltiazem, and verapamil for rhythm and rate control.
DIGOXIN TOXICITY
Over the past few decades the incidence and severity of digoxin toxicity has drastically decreased. A prospective study from 1971 noted toxic manifestations in as many as 24 percent of digoxin treated patients, with fatal outcomes in 41 percent of toxicity cases.48 More recent data suggest that 4–5 percent of digoxin treated patients develop toxicity, with fatality occurring in 9 percent of toxicity cases.49 Authors have speculated that this reduction in the incidence and severity of digoxin toxicity is related to decreasing utilization, lower dosing, and improved therapeutic drug monitoring.50 Despite these improvements toxicity and mortality secondary to toxicity still occurs. Therefore, it is important that clinical pharmacists understand the etiology, clinical manifestations, and management of digoxin toxicity.
A serum digoxin concentration (SDC) between 0.5 and 2.0 ng/mL is traditionally considered a therapeutic value, though some evidence suggests that lower levels may be equally as effective and reduce the risk of toxicity.50 Concentrations greater than 2.0 ng/mL are potentially toxic, with one review noting toxic manifestations in 60 percent of patients with a postdistribution SDC greater than 2.0 ng/mL.49 This finding illustrates that digoxin toxicity is at least in part concentration dependent and situations that precipitate an increase in concentration could also precipitate toxicity.
Digoxin is widely distributed and has an average VD of approximately 7 L/kg.5 Reductions in digoxin’s VD have been noted with renal disease and reductions in lean body mass.5,25 A number of factors, including renal disease and drug interactions, can decrease digoxin clearance (CL).5,25,50 Digoxin is mostly eliminated unchanged in the urine.5,25 Renal insufficiency reduces the elimination of digoxin and can significantly prolong digoxin’s half-life.5,50 A number of medications, including amiodarone, quinidine, and verapamil, reduce digoxin elimination via inhibition of the p-glycoprotein efflux pump.50 Reductions in CL will increase the apparent half-life with resulting drug accumulation with repeated dosing. Reductions in VD and CL will reciprocally increase SDCs and may lead to toxicity. Geriatric patients often are prescribed digoxin and are at greater risk for chronic poisoning.25 Reductions in lean body mass and declining renal function commonly occur with the natural aging course.25 These patients are also more commonly on multiple medications, putting them at an increased risk for toxicity secondary to drug interactions.
Clinical manifestations of digoxin toxicity are listed in Table 6-2. These signs and symptoms can be classified as gastrointestinal, neurologic, or cardiac manifestations, with cardiac manifestations being the primary cause of morbidity and mortality. Symptom evolution is dependent on the chronicity of exposure.51 Nausea and vomiting are prominent with acute exposures and are typically the first symptom.51 Neurologic symptoms such as weakness, confusion, and lethargy can be observed.52 With chronic exposures, such as a patient with reduced digoxin CL due to a physiologic change, symptoms can be nonspecific and more difficult to diagnose.51 Malaise and weakness are predominant features, along with visual disturbances, such as blurred vision.51-52 Patients may have complaints of anorexia, while nausea and vomiting are thought to occur less commonly than acute ingestions.
| TABLE 6-2 | Clinical Manifestations of Acute and Chronic Digoxin Toxicity |
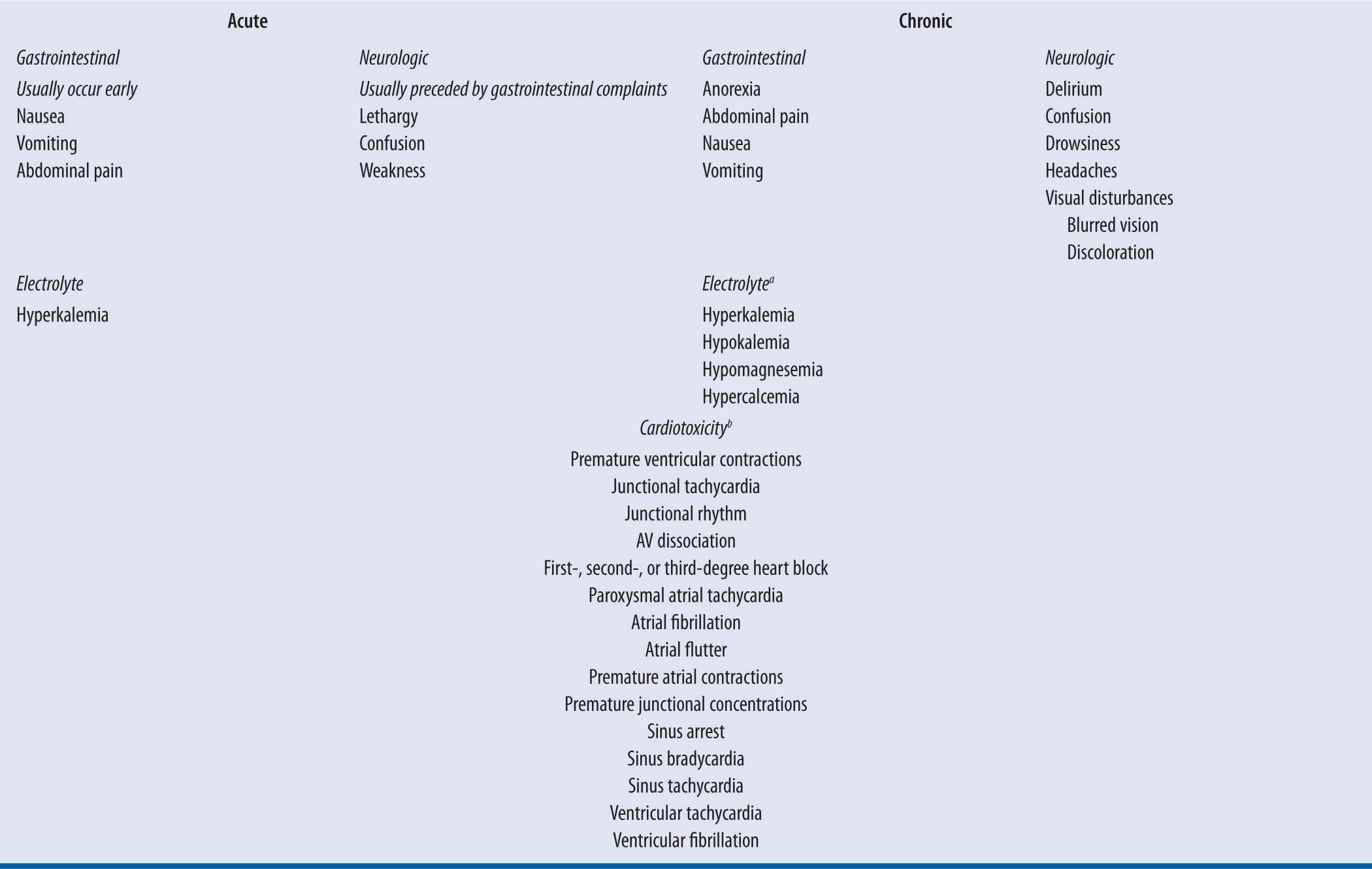
aThese electrolyte abnormalities can be seen with chronic exposures. They are not due to digoxin effect but due to concurrent medications such as diuretics or disease states such as renal failure. These abnormalities can exacerbate digoxin toxicity.
bCardiotoxicity can occur with both acute and chronic toxicity. Cardiac manifestations can be seen at presentation with chronic exposures. Conversely, with acute exposures these effects can be absent at presentation and they can progress as the clinical course evolves.
Digoxin’s cardiotoxicity is multifaceted and can be seen with both acute and chronic toxicity. At toxic levels digoxin increases intracellular calcium concentrations escalating excitability and automaticity in the atria and ventricles, which can lead to extra-systoles and tachydysrhythmias.50-51 Additionally, digoxin reduces nodal conduction velocity and can induce bradydysrhythmias and nodal blocks with toxicity.51 Thus, with the exception of a supraventricular tachycardia with rapid ventricular response, digoxin toxicity can precipitate almost any arrhythmia, with premature ventricular contractions being the most common.50-51 Bidirectional ventricular tachycardia is considered a pathognomonic finding, as it is uncommon for almost all other toxins.51 Finally, digoxin achieves its therapeutic effect by inhibiting myocardial sodium-potassium ATPase, increasing extracellular potassium concentrations.50 Hyperkalemia can occur, typically with acute overdoses, and is considered a prognostic factor. Prior to the advent of digoxin-specific Fab, death occurred in 50 percent and 100 percent of acutely poisoned digoxin patients with serum potassium levels of 5.0–5.5 mEq/L and >5.5 mEq/L, respectively.54 Additionally, patients chronically on digoxin are frequently on medications such as loop diuretics, which can exacerbate digoxin toxicity and cause other electrolyte abnormalities, such as hypokalemia and hypomagnesemia.55 Hypokalemia, like digoxin inhibits myocardial sodium-potassium ATPase and can independently precipitate arrhythmias. Therefore, hypokalemia could precipitate toxicity at lower and sometimes therapeutic digoxin concentrations.53
Digoxin follows a two-compartment model with compartments displaying rapid and slow distribution.5 SDCs are sampled from the rapidly distributing compartment. Conversely, the myocardium is found within the slowly distributing portion. The rapid and slowly distributing compartments do not reach distribution equilibrium until approximately 4 hours and 6 hours after the last administered intravenous or oral dose, respectively.55-56 Predistribution values can be falsely elevated, making interpretation difficult. One review of toxic levels (SDC >2 ng/mL) noted that 16 percent of toxic specimens were drawn during the predistribution period.49 When assessing a toxic SDC it is important to assess this value in the context of a patient’s symptoms and the timing of the level in relation to the last administered digoxin dose.
Digoxin-specific Fab (DSFab) is the definitive therapy for digoxin toxicity and is indicated with life-threatening or potentially life-threatening toxicity (see Table 6-3).57-58 Digibind® and DigiFab® are the commercially available preparations of DSFab, and the pharmacokinetic properties of these products are detailed in Table 6-4.57-58
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


