arise late in gestation and produce more confined defects (6). By contrast, a polytopic field defect is thought to result from an earlier defect during blastogenesis—the first 4 weeks of development—and occurs if abnormal inductive processes produce more distantly located and diverse defects (6,7).
When the etiology is known and easily remembered, the appropriate term should be used to designate the disorder.
Time-honored designations should be continued unless there is good reason to change.
In the absence of a reasonably descriptive designation, eponyms, some of them multiple, may be used until the basic defect for the disorder is recognized. However, use of an eponym should thereafter be limited to one proper name.
The use of the possessive form of an eponym should be discontinued, because the author neither had nor owned the disorder.
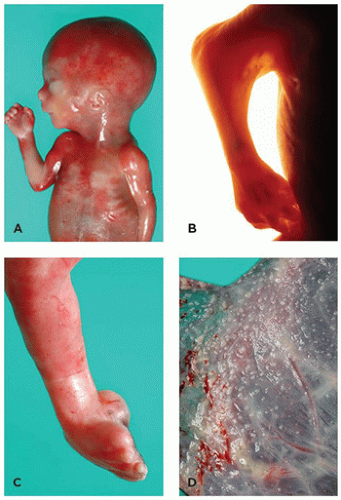
FIGURE 4-1 • Potter sequence. A: A 22-week fetus with a history of severe oligohydramnios (renal system normal; no history of premature rupture of membranes). Note the blunt nose, small mandible, and flattened ear. B: Marked skin webbing (pterygium) of right elbow developed secondary to prolonged immobilization of joint. C: Medial rotation of foot at ankle joint (talipes equinovarus) resulted from intrauterine constraint. D: Fetal surface of placenta shows amnion nodosum, a finding common in cases of oligohydramnios.
Designation of a disorder by one or more of its manifestations does not necessarily imply that they are either specific or consistent components of that disorder.
Names that may have an unpleasant connotation for the affected individual or family should be avoided.
The syndrome should not be designated by the initials of the originally described patients.
Names that are too general for a specific syndrome should be avoided.
Unless acronyms are extremely pertinent or appropriate, they should be avoided.
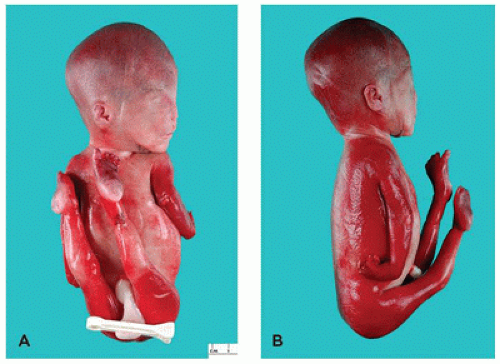 FIGURE 4-2 • Severe arthrogryposis in a 23-week fetus. Extraordinary flexion and contracture deformities and marked flattening of the face are apparent. Autopsy revealed no other fetal anomalies (karyotype 46,XX). Etiology is heterogeneous in this condition. A: Frontal view. B: Lateral view. |
that pregnant women should avoid all unnecessary radiation exposure. However, data regarding exact doses of radiation are often unavailable, and so actions based upon those fears (i.e., elective abortion after an exposure or suspected exposure) may be unwarranted. Counselors must use extreme caution when dealing with questions regarding radiation exposure.
of leprosy and access to the drug. The mechanism of action continues to be studied. The drug has anti-inflammatory properties, and some have suggested that defective angiogenesis in developing limb buds may also be operational (19).
TABLE 4-1 TERATOGENIC AGENTS IN HUMANS | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
TABLE 4-2 CRITICAL PERIODS IN HUMAN TERATOGENICITY | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
is associated with a host of anomalies, including microcephaly, trigonocephaly, porencephaly, spina bifida, other CNS defects, facial anomalies, cardiac defects, limb reduction anomalies, and genitourinary defects (33). Dosages associated with malformations have generally been 750 to 1,000 mg/day (although low-dose effects are also suspected) and exposures verified during the first trimester.
former has suggested a genetic influence (56). Despite small head circumference and initially slow psychomotor maturation, some infants with FAS may progress and develop intelligence within the normal range. Endocrine investigations usually show normal or near-normal levels of growth hormone, cortisol, and gonadotropins (see Chapter 21) (57). FAS is also a carcinogenic syndrome and is associated with tumors virtually identical to those seen in the fetal diphenylhydantoin (Dilantin) syndrome (see below).
TABLE 4-3 CHARACTERISTICS OF FAS | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
TABLE 4-4 DISORDERS ASSOCIATED WITH NUCHAL CYSTIC HYGROMA | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
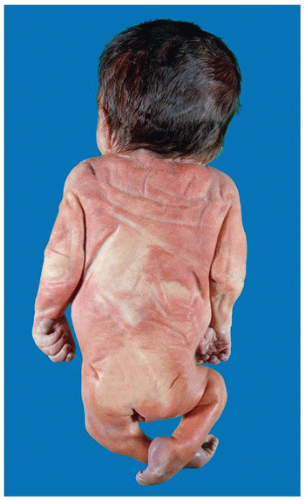 FIGURE 4-3 • Infant of diabetic mother. Pelvic girdle is reduced noticeably in this 31-week-old male with absent lumbosacral spine and malformed pelvis, indicative of caudal dysgenesis (formerly caudal regression syndrome). |
defect, varicella, Venezuelan equine encephalitis, Coxsackie virus, and syphilis. Acquired immune deficiency syndrome (AIDS) is acquired by transplacental means or during labor, delivery, or breast feeding and constitutes an enormous problem worldwide (72). In 2005 in the United States, 92% of cases of children with AIDS were attributed to maternal transmission of the human immunodeficiency virus (HIV); the incidence of neonatal HIV infection has fallen substantially in the United States with the implementation of prenatal testing, antiretroviral therapy, C-section, and avoidance of breast feeding (73).
TABLE 4-5 CONGENITAL ANOMALIES ASSOCIATED WITH MATERNAL DIABETES MELLITUS | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
the layer of skin affected. Secondary thrombosis with subsequent localized atrophy and ulceration may occur. Cutis marmorata telangiectatica congenita occurs sporadically, with female preponderance and occasional minor manifestations in close relatives.
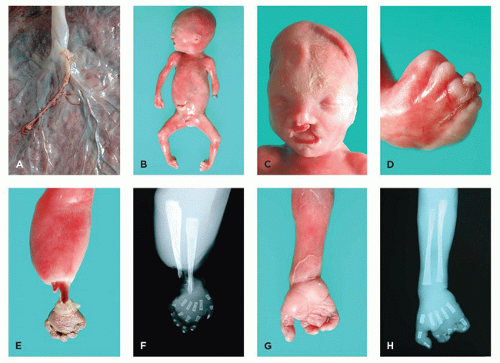 FIGURE 4-4 • Amnion rupture sequence. A: Close-up view of fetal surface of placenta shows tiny remnant of amnion. B: A 22-week male fetus with multiple amputation defects. C: Face with unilateral cleft lip. D: Right foot with syndactyly and multiple amputations of the digits. E: Exposed radius and ulna and necrosis of hand reflect the evolution of a band-induced amputation. F: Radiograph corresponding to (E). G: Right hand with multiple amputation defects. H: Radiograph corresponding to (G). |
TABLE 4-6 TIMING OF ANOMALIES ASSOCIATED WITH EARLY AMNION RUPTURE | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
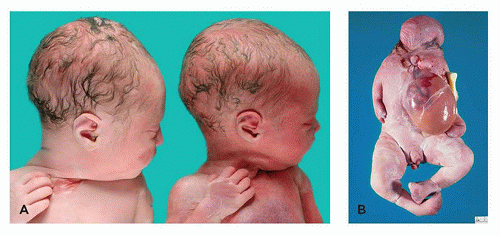 FIGURE 4-5 • Complications of monochorionic twinning. A: Pale, donor twin (left) and congested, recipient cotwin (right) in twin-twin transfusion syndrome. B: Acardiac cotwin in TRAP. Note the absence or malformation of structures of the upper body, omphalocele, and more normal lower extremities (but with anomalies of numerous digits). |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


