Chapter 35 Loralie Langman, Ph.D., F.C.A.C.B., D.A.B.C.C.(C.C.,M.B., T.C.), D.A.B.F.T., Laura Bechtel, Ph.D., D.A.B.C.C. and Christopher P. Holstege, M.D. Toxicology is a broad, multidisciplinary science whose goal is to determine the effects of chemical agents on living systems. Innumerable potential toxins can inflict harm, including pharmaceuticals, herbals, household products, environmental agents, occupational chemicals, drugs of abuse, and chemical terrorism threats. Each year millions of human exposure cases are reported to the American Association of Poison Control Centers.96 The Centers for Disease Control has reported that poisoning (both intentional and unintentional) is one of the top 10 causes of injury-related death in the United States in all adult age groups. From the beginnings of written history, poisons and their effects have been well described. Paracelsus (1493-1541) correctly noted that “Alle Ding sind Gift, und nichts ohn Gift; allein die Dosis macht, daß ein Ding kein Gift ist,” which means, “Everything is a poison; there is nothing which is not. The dose differentiates a poison.” As life in the modern era has become more complex, so has the study of poisons and their treatments. In practice, it is neither possible nor necessary to test for all of the hundreds or thousands of clinical toxins that may be encountered. In reality up to 24 drugs or agents account for 80% or more of cases of intoxication treated in most emergency departments.588 Moreover, some drugs are encountered very infrequently in some locations but with relatively high frequency in others. For example, phencyclidine (PCP) use is almost nonexistent in some areas but is responsible for a relatively high number of intoxications in a few large metropolitan cities. Thus the scope of clinical toxicology testing provided by the laboratory will depend on the pattern of local drug use and on the available resources of the institution and should be developed in consultation with the appropriate clinical staff. The value of drug and/or substance testing (screening) is well established (1) in the workplace, (2) for some athletic competitions, (3) to monitor drug use during pregnancy, (4) to evaluate drug exposure and/or withdrawal in newborns, (5) to monitor patients in pain management and drug abuse treatment programs, and (6) to aid in the prompt diagnosis of toxicity for a select number of drugs or agents for which a specific antidote or treatment modality is required (Table 35-1). In many other instances of drug toxicity, the value of drug screening, especially on an emergency basis, is more controversial.94,411,552,695 TABLE 35-1 Antidote or Specific Treatment for Intoxication 1. The time and date of the suspected exposure along with the time and date of sample collection. 2. History from the patient or witnesses that might aid in identification of the toxin. 3. Assessment of the physical state of the patient at the time of presentation. Such information is useful to guide test selection and interpretation of results. Because of the wide range of drugs of interest, no single analytical technique is adequate for broad-spectrum drug detection. Therefore several analytical approaches in combination are generally required. These may include simple, inexpensive, and rapid spot tests; immunoassays (see Chapter 16); and chromatographic and/or mass spectrometric techniques (see Chapters 13 and 14), including thin-layer chromatography (TLC), high-performance liquid chromatography (HPLC), gas chromatography (GC), gas chromatography-mass spectrometry (GC-MS or GC-MS/MS), and liquid chromatography-mass spectrometry (LC-MS or LC-MS/MS).772 Currently, GC-MS is the most widely used definitive confirmatory procedure, although LC-MS/MS is becoming more popular. Confirmatory testing is mandatory for forensic drug testing (e.g., workplace drug testing). Speed of analysis, or turnaround time (TOT), and availability are critical issues in clinical toxicology. A drug analysis that requires several hours to complete or that is not available at all hours of the day is of little value in a clinical emergency. Alternatively, a rapid test that provides false information could result in erroneous diagnostic and therapeutic decisions. For numerous agents, quantitative determinations guide management during a clinical emergency. These agents include (1) acetaminophen, (2) carbamazepine, (3) digoxin, (4) ethanol, (5) ethylene glycol, (6) iron, (7) isopropanol, (8) lithium, (9) methanol, (10) phenobarbital, (11) phenytoin, (12) salicylate, (13) valproic acid, and (14) theophylline, and in whole blood, (15) carboxyhemoglobin and (16) methemoglobin. Results for these determinations should be available within 1 hour of specimen receipt.860 When a healthcare team initially evaluates a patient who presents with a potential toxicologically induced health problem, the final diagnosis is often determined by (1) reviewing the history, (2) performing a directed physical examination, (3) using ancillary tests (e.g., electrocardiogram, radiology), and (4) applying a rational approach to laboratory testing. Often no specific antidote or treatment is available for a poisoned patient and careful supportive care is the most important intervention.234 Too often healthcare providers are lulled into a false sense of security when a patient presents with altered mental status and oxygen saturations on pulse oximetry that are adequate on high-flow oxygen. If the patient has inadequate ventilation or a poor gag reflex, then the patient may be at risk for progressive CO2 narcosis or aspiration, respectively, and yet may be maintaining adequate oxygen saturation on supplemental oxygen. It is imperative that the healthcare team avoid common complications of poisonings such as aspiration pneumonitis.138,359 An arterial blood gas (ABG) can rapidly aid the healthcare team in determining the need for intubation and mechanical ventilation. The ABG can also provide valuable information regarding the patient’s acid-base status and can help the clinician begin to generate a differential diagnosis. For example, in the scenario of a febrile toxic patient who presents with an altered mental status, a normal ABG (lack of acidosis) immediately rules-out uncoupling of oxidative phosphorylation as a cause of that patient’s fever. Finally, an ABG with co-oximetry can rapidly assist in determining other toxic etiologies, such as carbon monoxide poisoning (depending on the timing of the blood draw in relation to the exposure) and methemoglobinemia. The initial treatment of hypotension in all toxic patients consists of the administration of intravenous fluids.340 The patient’s pulmonary status should be closely monitored to ensure that pulmonary edema does not develop as fluids are infused. Symptomatic toxic patients should be placed on continuous cardiac monitoring with pulse oximetry, and the healthcare team must perform frequent neurologic checks to ensure continued protection of the airway. Acutely poisoned patients should receive a large-bore peripheral intravenous line, and all symptomatic patients should have a second line placed in the peripheral or central venous system, depending on the severity of their clinical status. At this time in patient care, blood can be drawn and sent for appropriate laboratory diagnostic testing. Placement of a urinary catheter should be considered early in the care of hemodynamically unstable poisoned patients to monitor urinary output as an indicator of adequate perfusion. A rapid bedside urine dipstick (photometric chemical assay) provides helpful information rapidly as healthcare team members await further laboratory testing. For example, a urine specific gravity will give insight into the patient’s initial hydration status, and the appearance of tea-colored urine positive for blood may indicate the presence of myoglobinuria in a comatose patient with rhabdomyolysis. Upon completion of the primary survey, the healthcare team should be assured that the patient’s airway is open and unblocked (also described as a patent airway), the ventilatory effort is adequate, and blood pressure is sustained in an appropriate range. At this point in management, the secondary survey can be performed. The secondary survey involves a thorough examination of the entire patient. For adequate access to a toxic patient, the patient must be completely undressed. Exposure of the patient ensures that a complete physical examination is performed.234 If the patient is not completely undressed, an important diagnostic clue may be missed. For example, skin lesions consistent with pressure necrosis on the back of a comatose patient may indicate the need to obtain a blood creatine phosphokinase activity or a urine myoglobin concentration. A comatose drug abuser may have attached transdermal drug patches (e.g., fentanyl, clonidine) in atypical locations (e.g., gluteal sulcus) that when found can rapidly lead to a diagnosis, avoiding the need for further laboratory testing. Besides completing a thorough physical review of all organ systems, the secondary survey involves reviewing items brought with the patient (e.g., medication bottles, drug paraphernalia). Searching carefully through the patient’s clothing may assist in providing clues that change the plan for specific laboratory tests or explain specific laboratory findings. For example, the discovery of a cough and cold product in the patient’s pocket that contains dextromethorphan could explain the clinical presentation of an agitated patient with hyperreflexia whose initial urine toxicology screen was positive for phencyclidine but later was found negative on confirmation. Toxic syndromes (“toxidromes”) are clinical syndromes that are essential for the successful recognition of poisoning patterns. A toxidrome is the constellation of clinical signs and symptoms that suggests a specific class of poisoning. An important component of the secondary survey is to determine whether a specific toxic syndrome is present.234 The most commonly encountered toxidromes include (1) anticholinergic, (2) cholinergic, (3) opioid, (4) sedative-hypnotic, and (5) sympathomimetic (Table 35-2). Many toxidromes have several overlapping features. For example, anticholinergic findings are highly similar to sympathomimetic findings, with one exception being the effects on sweat glands: anticholinergic agents produce warm, flushed dry skin, but sympathomimetic agents produce diaphoresis. Toxidrome findings may also be affected by individual variability, comorbid conditions, and coingestants. For example, tachycardia associated with sympathomimetic or anticholinergic toxidromes may be absent in a patient who is concurrently taking beta-antagonist medications. Additionally, although toxidromes may be applied to classes of drugs, one or more toxidrome findings may be absent for some individual agents within these classes. For instance, meperidine is an opioid analgesic, but it does not induce miosis, which helps to define the “classic” opioid toxidrome. When accurately identified, the toxidrome may provide invaluable information for diagnosis and subsequent treatment, although the many limitations impeding acute toxidrome diagnosis must be carefully considered. The respiratory effects of cholinergic poisoning tend to be dramatic and are considered to be the major factor leading to the death of its victims. Respiratory failure typically occurs as a triad of increased airway resistance, neuromuscular failure, and depression of central respiratory centers. Profuse watery nasal discharge, marked salivation, bronchorrhea, and bronchoconstriction result in a prolonged expiratory phase, cough, and wheezing. Because of the widespread presence of cholinergic receptors in the brain, cholinergic poisoning can produce great variation in neurologic signs and symptoms, including centrally mediated respiratory failure, coma, and seizures. Cholinergic cardiotoxicity can result in two clinical scenarios: a period of intense sympathetic activity that results in sinus tachydysrhythmias, or a period of increased parasympathetic tone that leads to bradydysrhythmias, prolongation of the PR interval, and atrioventricular block. Muscular symptoms may be vague and may consist of muscular weakness and difficulty with ambulation that can progress to muscular fasciculations and subsequent paralysis. Cholinergic agents cause constriction of both the sphincter muscle of the iris and the ciliary muscle of the lens, as well as stimulation of the lacrimal gland, resulting in lacrimation and miosis. The dermal sweat glands are innervated by sympathetic muscarinic receptors. When these receptors are stimulated, profuse sweating occurs. Cholinergic gastrointestinal and genitourinary symptoms may result in nausea, vomiting, abdominal cramps, tenesmus, and involuntary defecation and urination. Two mnemonics have been developed to help recall cholinergic clinical effects: DUMB BELS (diarrhea, urination, miosis, bradycardia, and bronchorrhea-bronchoconstriction, emesis, lacrimation, sweating-salivation)415 and SLUDGE (salivation, lacrimation, urination, defecation, GI distress, emesis-eye findings, miosis).145 Toxin-induced hyperthermic syndromes are potentially devastating and require rapid management. Even though the patient’s temperature is one of the vital signs, the temperature often is not obtained in clinical practice. Fever in a poisoned patient can be associated with several hyperthermic syndromes: sympathomimetic toxicity, uncoupling of oxidative phosphorylation, serotonin syndrome, neuroleptic malignant syndrome, malignant hyperthermia, anticholinergic poisoning, and withdrawal syndromes.692 Sympathomimetics, such as amphetamines and cocaine, may produce hyperthermia as the result of excess serotonin and dopamine, leading to thermal deregulation.372 Uncoupling of oxidative phosphorylation, as seen in severe salicylate poisoning, occurs when the process of oxidative phosphorylation is disrupted, leading to heat generation and a reduced ability to aerobically generate adenosine-5′-triphosphate (ATP).409 Serotonin syndrome occurs when a relative excess of serotonin is present at both peripheral and central serotonergic receptors.360 Patients may present with hyperthermia, alterations in mental status, and neuromuscular abnormalities (rigidity, hyperreflexia, clonus), although individual variability is noted in these findings. Serotonin syndrome is associated with drug interactions such as those associated with the combination of monoamine oxidase inhibitors and meperidine, but it may also occur with single-agent therapeutic dosing or overdosing of serotonergic agents. Neuroleptic malignant syndrome is a condition caused by relative deficiency of dopamine within the central nervous system.717 It has been associated with dopamine receptor antagonists and the withdrawal of dopamine agonists such as levodopa/carbidopa products. Clinically, it may be difficult to distinguish from serotonin syndrome and other hyperthermic emergencies. Malignant hyperthermia occurs when genetically susceptible individuals are exposed to depolarizing neuromuscular blocking agents or volatile general anesthetics. Anticholinergic poisoning may result in hyperthermia through impairment of normal cooling mechanisms such as sweating. Withdrawal syndromes can produce excessive adrenergic responses (e.g., sedative-hypnotic withdrawal, ethanol withdrawal) and subsequent heat generation. It should be noted that opioid withdrawal is not associated with fever or altered mental status. Overall, differentiating between the toxic hyperthermic syndromes may be challenging, and additional causes of hyperthermia such as heat stroke and infection should be explored. Two bedside tests have been proposed for rapid identification of the patient with salicylate toxicity. Both involve simply applying a few drops of a prepared reagent to a small sample of a patient’s urine and watching for a characteristic color change. The first such reagent is ferric chloride (FeCl3). Applying a few drops of a 10% solution of ferric chloride to 1 mL of urine containing even very small amounts of salicylate will produce a characteristic purple color caused by the formation of an iron-salicylate complex.337 This color change also will occur if the urine in question contains acetoacetic acid and phenylpyruvic acid.250 The urine Trinder spot test uses a reagent composed of mercuric chloride, ferric nitrate, concentrated hydrogen chloride, and deionized water.421 Applying 1 mL of this solution to 1 mL of urine with salicylates present will also lead to a purple color change.250 One study exploring the salicylate question254 examined the use of the ferric chloride test in 187 patients presenting to the emergency department. These ferric chloride tests were subsequently followed with serum salicylate concentrations. The sensitivity of the ferric chloride was 93.2% for salicylate concentrations ≥3.0 mg/dL and 93.8% for concentrations ≥30.0 mg/dL. Specificity was 88.8% and 75.4%, respectively. Three false negatives were reported, one of which had a toxic salicylate concentration of 34 mg/dL. King and associates evaluated the clinical utility of the Trinder reagent.421 Investigators enlisted 12 volunteers who ingested 975 mg of aspirin. The sensitivity of the test in this study was 100% with two false positives from controls. Weiner and colleagues examined the application of these two bedside tests in patients presenting to the emergency department with suspected drug overdose with or without unexplained metabolic acidosis.827 Both reagents were added to urine samples from all 180 patients enrolled, and confirmatory serum salicylate concentrations were drawn. Different from the previous studies, however, any darkening of color was regarded as a positive test. Twenty of these patients (11%) had salicylate concentrations ≥5 mg/dL. Both tests were 100% sensitive for recognizing these patients. The specificities for both tests were relatively low (Trinder with 73% and ferric chloride with 71%). Automotive antifreeze is a major source of ethylene glycol exposure. Sodium fluorescein is added to the antifreeze to aid in identifying cooling system leaks. Some have suggested that fluorescein excreted in the urine of a patient who has ingested antifreeze would fluoresce with the aid of a Wood’s lamp.818 Unfortunately, it seems that little advantage is derived by checking for urinary fluorescence by Wood’s lamp in those suspected of ethylene glycol poisoning.120,818 Obtaining a basic metabolic panel in all poisoned patients is recommended and is an important initial screening test. When low serum bicarbonate is discovered on a metabolic panel, the clinician should determine whether an elevated anion gap exists. The formula most commonly used for the anion gap (AG) calculation is as follows124: This equation allows one to determine whether serum electroneutrality is being maintained. The primary cation (sodium) and anions (chloride and bicarbonate) are represented in the equation.364 Other contributors to this equation are “unmeasured.”267 Other serum cations are not commonly included in this calculation because either their concentrations are relatively low (e.g., potassium) or assigning a number to represent their respective contribution is difficult (e.g., magnesium, calcium).267 Similarly, a multitude of other serum anions (e.g., sulfate, phosphate, organic anions) are also difficult to measure and quantify in an equation.267,364 These “unmeasured” ions represent the anion gap calculated using the previous equation. The reference interval for this anion gap is accepted to be 8 to 16 mmol/L,267 but it has been suggested that because of changes in the technique used to measure chloride, the interval should be lowered to 6 to 14 mmol/L.364 Practically speaking, an increase in the anion gap beyond an accepted reference interval, accompanied by a metabolic acidosis, represents an increase in unmeasured endogenous (e.g., lactate) or exogenous (e.g., salicylates) anions.124 A list of the more common causes of this phenomenon is organized in the classic MUDILES pneumonic (Box 35-1). Note: The “P” has been removed from the older acronym of MUDPILES, because paraldehyde is no longer available. Interpretation of the electrocardiogram (ECG) in the poisoned patient can significantly facilitate appropriate laboratory testing, diagnosis, and management of the poisoned patient, because numerous drugs can cause ECG changes.832 Despite the fact that drugs have widely varying indications for therapeutic use, many unrelated drugs share common cardiac electrocardiographic effects if taken in overdose. Potential toxins can be placed into broad classes on the basis of their cardiac effects. For example, agents that block cardiac potassium efflux channels and agents that block cardiac fast sodium channels can lead to characteristic changes in cardiac indices consisting of QRS prolongation and QT prolongation, respectively. The recognition of specific ECG changes associated with other clinical data (toxidromes) can be potentially lifesaving.341 Radiologic testing is sometimes used to diagnose complications associated with poisonings, such as aspiration pneumonitis and anoxic brain injury. The use of radiology has also been advocated to detect the presence of potentially radiopaque poisons. For example, O’Brien and associates studied the detectability of 459 different tablets and capsules using plain radiography.596 Investigators used a ferrous sulfate tablet as a control in grading the radiopacity of other tablets. Overall, of the wide variety of pills tested, only 6.3% were graded as having radiopacity the same as or greater than ferrous sulfate; 29.6% were regarded as having at least moderate opacity; and the largest remaining portion of pills (64%) was regarded as no more than minimally detectable. Based on this and other studies, the indiscriminate use of plain abdominal x-rays is not justified, and a negative film should not be relied upon to rule out potential toxic pill ingestion, especially if enough time is given to allow the pills to dissolve. The main osmotically active constituents of serum are Na+, Cl−, Elevated OSMg implies the presence of unmeasured osmotically active substances.427 Volatile alcohols (ethanol, methanol, isopropanol, acetone, and ethylene glycol) when present at significant concentrations, increase serum osmolality, thus resulting in an increased OSMg. The calculation of OSMg is commonly used as a screen.276 However, it is important to remember that volatile alcohols are not detected when osmolality is measured with a vapor pressure osmometer. Therefore, for the purpose of determining the OSMg, only osmolality measurements based on freezing-point depression are acceptable. What constitutes a normal osmol gap is widely debated. Traditionally, a normal gap has been defined as 10 mOsm/kg or less. The original source of this value is an article by Smithline and Gardner,742 which declared that this number was pure convention. Further clinical study has not shown this assumption to be correct. However, large variability is seen in the normal population.456,558 Researchers have found the OSMg to vary from −9 to +5 mOsm/kg288; from −13.5 to +8.9536; and from −10 to +20 mOsm/kg,1 depending on the population studied. An important point to consider is that the day-to-day coefficient of variance of sodium was 1%. This analytical variance alone may account for the variation found in patients’ osmol gaps.1 One would expect that each 100 mg/dL (21.7 mmol/L) of ethanol (molecular weight = 46.068 g/mol) in serum results in an approximate increase of 21.7 mOsm/kg.640 However, this is not found to be the case. Applying a correction factor of 0.83 to the ethanol value will more closely approximate the contribution of ethanol to the OSMg.276 By considering this effect of ethanol on the serum osmolality, it is possible to determine what portion of an increased osmol gap is due to ethanol. The contribution of ethanol to the measured osmolality can be calculated (ethanol, mg/dL/4.6 × 0.83) and included in the preceding formula for delta osmolality calculation. However, it has been observed that ethanol and methanol do not follow a completely predictable relationship with OSMg. In severe ethanol and methanol intoxication, OSMg increases with increasing concentration, making it appear that something is present besides the alcohol.276,469,539,606,662 A significant residual osmol gap (>10 mOsm/kg) after the correction for ethanol would suggest the possible presence of isopropanol, methanol, acetone, or ethylene glycol. This information, in conjunction with the presence or absence of metabolic acidosis or serum acetone, is helpful to the clinician when specific measurements of alcohols other than ethanol and of ethylene glycol are not available on an emergency basis (Table 35-3). It must be realized that ketones (diabetics) and substances administered to patients such as polyethylene glycol (burn cream),98 mannitol (osmotic diuretic), and propylene glycol (solvent for diazepam and phenytoin) may increase serum osmolality. For the diagnosis of ethanol intoxication, OSMg poisoning has lost its usefulness because ethanol is measured quickly on most chemistry analyzers. However, because other toxic alcohols can be measured only by chromatographic techniques, it is still useful. Unfortunately, OSMg as a screening method is insensitive to low, yet clinically significant, concentrations of ethylene glycol (<50 mg/dL), and methanol (<30 mg/dL).616 Different types of immunoassays are useful in screening specimens for drugs (see Chapters 16 and 34). In some cases, these assays are relatively specific for a single drug (LSD), but in others, several drugs of a similar class are detected (e.g., opiates). The detection limit for various members of a class of drugs or the degree of cross-reactivity for similar drugs varies, and each manufacturer of immunoassay reagents should be consulted for specific information. These assays are easy to perform; many are available for use on automated instrumentation and may be able to provide “semiquantitative” results. Several portable, noninstrumental, immunoassay-based drug detection devices are available for use in point-of-care testing (POCT). For the vast majority of drugs of abuse, immunoassays are the methods of choice for initial screening. However, for a more comprehensive drug screening, chromatographic procedures complement immunoassays. Planar chromatography, commonly known as thin-layer chromatography (TLC), is a versatile procedure that requires no instrumentation and thus is operationally relatively simple and inexpensive (see Chapter 13). With this technique, a large number of drugs may be detected; it may be applied to the analysis of serum, gastric contents, or urine. Urine, however, is the specimen of choice because most drugs and drug metabolites are present in urine in relatively high concentrations. However, application of TLC to drug screening requires considerable experience and skill to recognize drug and metabolite patterns and various color hues for detection; it has largely been replaced by other chromatographic techniques. For a more comprehensive description, refer to previous editions of this textbook.78 Also known as gas liquid chromatography, GC is relatively rapid, is capable of resolving a broad spectrum of drugs, and is widely used for qualitative and quantitative drug analysis.392 Capillary columns, because of their high efficiency, are analytical columns that are commonly used for drug detection by GC (see Chapter 13). In many instances, nonderivatized drugs have good GC properties when capillary columns are used; in some instances, derivatization to a less polar or more volatile compound is necessary. Common detectors for drug detection by GC are flame ionization and alkali flame ionization (nitrogen phosphorus) detectors and mass spectrometers, which provide the greatest accuracy of identification. Numerous methods for general drug screening by GC-MS spectrometry have been published, but one comprehensive method can be adapted to multiple body fluids and tissues.392 The resolving power of HPLC (see Chapter 13) for separating widely divergent chemical constituents has been applied to the complex challenge of comprehensive drug screening in biological fluids. Advantages of HPLC over GC include the ability to analyze polar and thermally labile drugs without derivatization. The advent of diode array detectors that provide a spectral scan of compounds as they elute from the column greatly increased the discriminatory power of this technique.82,500,661,785 LC-MS or LC-MS/MS currently plays a limited role in comprehensive screening but is rapidly gaining in popularity.* Numerous POC drug test devices for urine (and oral fluid) are designed for easy, rugged, and portable use by nontechnical personnel. Although these devices are relatively simple to use, proper training of nonlaboratory users is important for optimal performance (see Chapter 20).279,445 These noninstrumental immunoassay test devices are designed for use at the site of collection; results are available within minutes and are variously configured to detect only one drug or many drugs simultaneously. The spectrum of drugs tested includes tricyclic antidepressants, barbiturates, benzodiazepines, methadone, MDMA (methylenedioxymethamphetamine), MDA (methylenedioxyamphetamine), MDEA (methylenedioxyethylamphetamine), oxycodone, and the traditional SAMHSA (Substance Abuse and Mental Health Services Administration) or NIDA (National Institute on Drug Abuse) 5 (amphetamine, cocaine, marijuana, opiates, and phencyclidine). As previously cited, such devices are also available for measurement of acetaminophen and salicylate in serum or whole blood. Evaluations for some of these test devices for urine and oral fluid558,639 have been published. A comprehensive review of on-site drug testing is also available.629 The assay principles of these POC test devices include the following: • Sequential competitive binding microparticle capture immunoassay • Homogeneous microparticle capture immunochromatography • Solid-phase competitive sequential enzyme immunoassay A more detailed description of these methods can be found in Chapter 16 and in the package insert for each specific test kit. Carbon monoxide (CO) is a colorless, odorless, tasteless gas that is a product of incomplete combustion of carbonaceous material. Common exogenous sources of carbon monoxide include cigarette smoke, gasoline engines, and improperly ventilated home heating units. Small amounts of carbon monoxide are produced endogenously in the metabolic conversion of heme to biliverdin.458 This endogenous production of carbon monoxide is accelerated in hemolytic anemias.496 When inhaled, carbon monoxide combines tightly with the heme Fe2+ of hemoglobin to form carboxyhemoglobin. The binding affinity of hemoglobin for carbon monoxide is about 250 times greater than that for oxygen. Therefore high concentrations of carboxyhemoglobin limit the oxygen content of blood. Moreover, the binding of carbon monoxide to a hemoglobin subunit increases the oxygen affinity for the remaining subunits in the hemoglobin tetramer. Thus at a given tissue PO2 value, less oxygen dissociates from hemoglobin when carbon monoxide is also bound, shifting the hemoglobin-oxygen dissociation curve to the left. Consequently, carbon monoxide not only decreases the oxygen content of blood, it also decreases oxygen availability to tissue, thereby producing a greater degree of tissue hypoxia than would result from an equivalent reduction in oxyhemoglobin due to hypoxia alone.368,778 Carbon monoxide may also bind to other heme proteins, such as myoglobin and mitochondrial cytochrome oxidase a3; this may limit oxygen use when tissue PO2 is very low.368,778 The toxic effects of carbon monoxide are a result of hypoxia. Organs with high oxygen demand, such as heart and brain, are most sensitive to hypoxia and thus account for the major clinical sequelae of carbon monoxide poisoning. It must be emphasized that the carboxyhemoglobin concentration, although helpful in diagnosis, does not always correlate with the clinical findings or prognosis.569,702 Factors other than carboxyhemoglobin concentration that contribute to toxicity include length of exposure, metabolic activity, and underlying disease, especially cardiac or cerebrovascular disease. Moreover, low carboxyhemoglobin concentrations relative to the severity of poisoning may be observed if the patient was removed from the carbon monoxide–contaminated environment several hours before blood sampling.318 An insidious effect of carbon monoxide poisoning is the delayed development of neuropsychiatric sequelae, which may include personality changes, motor disturbances, and memory impairment. These manifestations do not correlate with the length of exposure or with the maximum blood carboxyhemoglobin concentration.45 Treatment for carbon monoxide poisoning involves removal of the individual from the contaminated area and administration of oxygen. The half-life (t1/2) of carboxyhemoglobin in the body is variable, and attempts to determine the exact elimination t1/2 for CO based on the inhaled oxygen concentration have not been validated. Hyperbaric oxygen therapy for CO is highly debated, and current position papers have found no evidence to support its use.101 Gas chromatographic methods measure the carbon monoxide content of blood. When blood is treated with potassium ferricyanide, carboxyhemoglobin is converted to methemoglobin, and carbon monoxide is released into the gas phase. Measurement of the released carbon monoxide may be performed by GC using a molecular sieve column and a thermal conductivity detector.213 A lower detection limit is achieved by incorporating a reducing catalyst (e.g., nickel) between the GC column and the detector to convert carbon monoxide to methane. The methane may then be detected with a flame ionization detector.307 A very low detection limit may be achieved with the use of a heated mercuric oxide reaction chamber between the GC column and an ultraviolet light detector. As carbon monoxide elutes from the column, it reacts with mercuric oxide to form mercury gas, which has a high molar absorptivity at 254 nm.809 In practice, the carbon monoxide binding capacity is also determined after an aliquot of the blood specimen is treated with carbon monoxide to saturate the hemoglobin. The results are then expressed as percent of carboxyhemoglobin: GC methods are accurate and precise and are considered to be reference procedures. Normal values for carboxyhemoglobin in rural nonsmokers are about 0.5%; for urban nonsmokers, 1 to 2%; and for smokers, 5 to 6%.49 Values may be increased by about 3% in cases of hemolytic anemia.496 Spectrophotometric methods rely on the characteristic spectral absorption properties of carboxyhemoglobin.197,879 Among several such methods, the most popular are based on automated, multiwavelength measurements of several hemoglobin species. These methods are rapid and convenient for the determination of carboxyhemoglobin and other hemoglobin species. Spectrophotometric methods generally compare favorably with gas chromatographic procedures at carboxyhemoglobin concentrations greater than 2 to 3%, but their precision is poor below these concentrations.809 Therefore, they are sufficiently accurate and precise for measurement of carbon monoxide after exogenous exposure but are too insensitive to detect the increased endogenous production of carbon monoxide that occurs in hemolytic anemia. Fetal hemoglobin has slightly different spectral properties than adult hemoglobin. Consequently, falsely high carboxyhemoglobin values of 4 to 7% may occur when blood from neonates is measured by some spectrophotometric methods utilizing fewer wavelengths.810 Moreover, erroneous results may occur with lipemic specimens, with bilirubin, and in the presence of methylene blue (see section on “Methemoglobin-Forming Agents”). Hydrocyanic acid binds to hemoglobin. The hydrocyanic acid bound in the erythrocyte is in equilibrium with free hydrocyanic acid in the serum at a ratio of 10 : 1. Cyanide in serum readily crosses all biological membranes and avidly binds to heme iron (Fe3+) in the cytochrome a-a3 complex within mitochondria.715,803 When bound to cytochrome a-a3, cyanide is a competitive inhibitor that causes decoupling of oxidative phosphorylation. Patients exposed to toxic concentrations of cyanide exhibit rapid onset of symptoms typical of cellular hypoxia—flushing, headache, tachypnea, dizziness, and respiratory depression—which progress rapidly to coma, seizures, complete heart block, and death if the dose is sufficiently large. Following microdiffusion, whole blood CN− is measured by photometric analysis179 or by headspace gas chromatography.111 With the spectrophotometric method, a sealed, two-well microdiffusion cell is used to separate hydrocyanic acid from blood by mixing a sample of whole blood with strong acid in a sealed chamber and allowing the hydrocyanic acid gas generated to be absorbed into a strong base located in another part of the sealed chamber. One well of the cell contains the blood specimen and strong acid (unmixed until the cell is sealed), and the other well contains a strong base to absorb the hydrocyanic acid gas. After the hydrocyanic acid is collected in the aqueous base medium, pyridine, barbituric acid, and chloramine-T are added to generate a red complex, with the intensity of the color proportional to the concentration of CN−. A good quality spectrophotometer is required to measure the absorbance. Quick and easy methods for plasma thiocyanate analysis have been described.802 The heme iron in hemoglobin is normally present in the ferrous state (Fe2+). When oxidized to the ferric state (Fe3+), methemoglobin is formed, and this form of hemoglobin cannot bind oxygen (Figure 35-1). The principal physiologic system that maintains hemoglobin iron in the reduced state is nicotinamide adenine dinucleotide (NADH)-methemoglobin reductase. The NADH for this enzyme is supplied by normal glycolysis (Embden-Meyerhof pathway). A minor pathway for methemoglobin reduction involves nicotinamide adenine dinucleotide phosphate (NADPH)-methemoglobin reductase, and the NADP for this enzyme reaction is derived from the hexose-monophosphate shunt. Congenital methemoglobinemia may result from a deficiency of NADH-methemoglobin reductase or, more rarely, from hemoglobin variants (hemoglobin M) in which heme iron is both more susceptible to oxidation and more resistant to reduction by the methemoglobin reductase system. An acquired (toxic) methemoglobinemia may be caused by various drugs and chemicals (Table 35-4). The normal percentage of methemoglobin is <1.5% of total hemoglobin. In otherwise healthy individuals, methemoglobin percentages up to 20% may cause slate-gray cutaneous discoloration, cyanosis, and chocolate-brown blood. Percentages between 20% and 50% may cause dyspnea, exercise intolerance, fatigue, weakness, and syncope. More severe symptoms of dysrhythmias, seizures, metabolic acidosis, and coma are associated with methemoglobin percentages of 50 to 70%, and >70% may be lethal.659,858 All of these symptoms are a consequence of hypoxia associated with the diminished O2 content of the blood, and with a decreased O2 dissociation from hemoglobin species in which some, but not all, subunits contain heme iron in the ferric state (i.e., shift of dissociation curve to the left). The PO2 is normal in these patients, and therefore so is the calculated hemoglobin oxygen saturation. Thus, a normal PO2 in a cyanotic patient is a significant indication for the possible presence of methemoglobinemia. Direct measurement of methemoglobin is important in these cases and may be performed by the manual spectrophotometric method of Evelyn and Malloy235 or by automated multiwavelength measurements with a co-oximeter (see section on “Carbon Monoxide”). TABLE 35-4 Examples of Acquired Causes of Methemoglobinemia Specific therapy for toxic methemoglobinemia involves the administration of methylene blue, which acts as an electron transfer agent in the NADPH-methemoglobin reductase reaction, thereby increasing the activity of this system several-fold.649,658 Methylene blue and sulfhemoglobin cause spectral interference in the measurement of methemoglobin with some co-oximeters412,875 but not with the Evelyn-Malloy method.412 Methemoglobin is measured in blood manually,235,412 or by automated multiwavelength measurements with a co-oximeter.325 Methemoglobin interferes with the noninvasive pulse oximetry method, measuring the absorbance of light at 660 nm (oxyhemoglobin) and 940 nm (deoxyhemoglobin). Because methemoglobin is not stable at room temperature, specimens should be kept on ice or refrigerated but not frozen.412 The stability of methemoglobin at 4 °C has not been well studied. Some sources indicate significant decreases in methemoglobin concentration after 4 to 8 hours,470 whereas others report little or no change after 24 hours.412 Freezing results in an increase in methemoglobin concentration.412 Several alcohols are toxic and medically important.197A They include ethanol, methanol, isopropanol, acetone, and ethylene glycol. The principal pharmacologic action of ethanol is central nervous system (CNS) depression. CNS effects vary depending on the blood ethanol concentration (Table 35-5) but are also heavily influenced by an individual’s tolerance. Symptoms vary from euphoria and decreased inhibitions, to increased disorientation and incoordination, and then to coma and death. A blood alcohol concentration of 80 mg/dL (0.08%) has been established as the per se limit for operation of a motor vehicle in most countries. TABLE 35-5 Stages of Acute Alcoholic Influence/Intoxication Modified from Dubowski KM, Gadsden RH Sr, Poklis A. The stability of ethanol in human whole blood controls: an interlaboratory evaluation. J Anal Toxicol. 1997 Oct;21(6):486-91. All rights reserved. Because of many factors, not all individuals experience the same degree of CNS dysfunction at similar blood alcohol concentrations. Moreover, the CNS actions of ethanol are more pronounced when the blood ethanol concentration is increasing (absorptive phase) than when it is declining (elimination phase), in part because of the phenomenon of acute tolerance.291 In addition, heavy alcohol use leads to a more chronic form of tolerance. When consumed with other CNS depressant drugs, ethanol exerts a potentiation or synergistic depressant effect. This can occur at relatively low alcohol concentrations, and numerous deaths have resulted from combined ethanol and drug ingestion.274 The pharmacologic mechanisms for the CNS depressant actions of ethanol are complex and incompletely understood, but probably involve both enhancement of major inhibitory neurons and impairment of excitatory neurons. The principal CNS inhibitory neuronal system is mediated by the neurotransmitter γ-aminobutyric acid (GABA). When GABA binds to its postsynaptic receptor subtype GABAA, this oligomeric ion-gated complex “opens” to allow inward flux of Cl, leading to membrane hyperpolarization and subsequent decreased electrical response. This GABA-mediated inhibitory response is enhanced by ethanol and sedative, hypnotic, and anesthetic agents, including barbiturates, benzodiazepines, and volatile anesthetics.248 Neuronal nicotinic acetylcholine receptors also may be prominent molecular targets of alcohol.576 Both enhancement and inhibition of nicotinic acetylcholine receptor function have been reported depending on receptor subunit concentration and the concentrations of ethanol tested. Ethanol also inhibits the function of the N-methyl-D-aspartate (NMDA)- and kainate-receptor subtypes; AMPA receptors are largely resistant to alcohol.119 The aforementioned chronic tolerance to ethanol is considered to be mediated by ethanol-induced increased responsiveness and upregulation in the synthesis of NMDA receptors, attained by concomitant downregulation and desensitization through phorphorylation of GABAA and glutamate receptors.248,452,819 Largely because of these adaptive changes, abrupt withdrawal from chronic, heavy ethanol use leads to a physical abstinence syndrome that has prominent features of CNS excitation. Included among these withdrawal symptoms are anxiety, irritability, insomnia, muscle tremor and cramps, seizures, hallucinations, and increased temperature, blood pressure, and heart rate. Ethanol is metabolized principally by liver alcohol dehydrogenase to acetaldehyde, which is subsequently oxidized to acetic acid by aldehyde dehydrogenase (Figure 35-2). The rate of elimination of ethanol from blood approximates a zero-order process. This rate varies among individuals, averaging about 15 mg/dL/h for males and 18 mg/dL/h for females.210,211 At both low (<20 mg/dL)813 and high (>300 mg/dL) ethanol concentrations, elimination becomes more nearly first-order; it is accelerated at high concentrations.81 The elimination rate is also influenced by drinking practices (e.g., alcoholics have increased elimination rates caused by enzyme induction).847 Ethanol is a teratogen, and alcohol consumption during pregnancy can result in the birth of a baby with fetal alcohol spectrum disorder (FASD). FASD is an umbrella term that describes the variety of effects that can occur in an individual whose mother drank alcohol during pregnancy (http://www.nofas.org/accessed June 13, 2011). These effects may include physical, mental, behavioral, and/or learning disabilities with possible lifelong implications and are 100% preventable when a woman completely abstains from alcohol during her pregnancy. The CNS effects of methanol are substantially less severe than those of ethanol. Methanol is oxidized by liver alcohol dehydrogenase (at about one tenth the rate of ethanol) to formaldehyde. Formaldehyde in turn is rapidly oxidized by aldehyde dehydrogenase to formic acid, which may cause serious acidosis and optic neuropathy, resulting in blindness or death.513,534 Serum formate concentrations correlate better with the degree of acidosis and the severity of CNS and ocular toxicity than do serum methanol concentrations.718 Therefore, some investigators recommend the measurement of serum formate to assess the severity of toxicity and to guide appropriate therapy in cases of methanol ingestion. The mainstay of therapy for methanol toxicity includes the administration of ethanol or fomepizole as a competitive alcohol dehydrogenase inhibitor, either folate or folinic acid, and dialysis. Isopropanol is readily available to the general population as a 70% aqueous solution for use as rubbing alcohol. It has about twice the CNS depressant action as ethanol, but it is not as toxic as methanol.58 Isopropanol has a short t1/2 of 2.5 to 3.0 hours,58 as it is rapidly metabolized by alcohol dehydrogenase to acetone, which is eliminated much more slowly (t1/2, 3 to 6 hours).47 Therefore concentrations of acetone in serum often exceed those of isopropanol during the elimination phase following isopropanol ingestion. Acetone has CNS depressant activity similar to that of ethanol, and because of its longer t1/2, it may prolong the apparent CNS effects of isopropanol.47,58 Supportive care is the mainstay of treatment, with rare reports of dialysis in severe intoxication. Ethylene glycol itself is relatively nontoxic, and its initial CNS effects resemble those of ethanol.386 However, metabolism of ethylene glycol by alcohol dehydrogenase (ADH) results in the formation of numerous acid metabolites, including lactate, oxalic acid and glycolic acid.53,386 These acid metabolites are responsible for much of the toxicity of ethylene glycol.334,371 Serum concentrations associated with death from ethylene glycol ingestion have been observed to vary from 0.06 to 4.3 g/L,53,651 highlighting the lack of correlation between ethylene glycol concentration and severity of toxicity. It is thus impossible to define a serum ethylene glycol concentration associated with a high probability of death. The serum concentration of glycolic acid correlates more closely with clinical symptoms and mortality than does the concentration of ethylene glycol.334,651 Because of the rapid elimination of ethylene glycol (t1/2, 2 to 5 hours),53 its serum concentration may be low or undetectable at a time when glycolic acid remains elevated.257,334,651 Thus the determination of ethylene glycol and glycolic acid provides useful clinical and confirmatory analytical information in cases of ethylene glycol ingestion. The mainstay of therapy for ethylene glycol toxicity includes administration of ethanol or fomepizole as a competitive alcohol dehydrogenase inhibitor and dialysis. Alcohol distributes into the aqueous compartments of blood; because the water content of serum is greater than that of whole blood, higher alcohol concentrations are obtained with serum as compared with whole blood. Experimentally, the serum-to-whole blood ethanol ratio is 1.18 (1.10 to 1.35)628 and varies slightly with hematocrit.846 Therefore, laboratories that perform alcohol determinations should make clear the choice of specimen. Because of the volatile nature of alcohols, specimens should be kept capped to avoid evaporative loss. Blood may be stored, when properly sealed, for 14 days at room temperature or at 4 °C, with or without preservative.848 For longer storage or for nonsterile postmortem specimens, sodium fluoride should be used as a preservative to prevent a decrease or occasionally an increase (via fermentation) in ethanol concentration. To measure ethanol in serum/plasma, enzymatic analysis is the method of choice for many laboratories. In this method, ethanol is measured by oxidation to acetaldehyde with NAD, a reaction catalyzed by ADH. With this reaction, the formation of NADH, measured at 340 nm, is proportional to the amount of ethanol in the specimen268: Under most assay conditions, ADH is reasonably specific for ethanol, with interferences by isopropanol, acetone, methanol, and ethylene glycol of typically <1%. However, spuriously increased results for ethanol have been described in the presence of high concentrations of lactate dehydrogenase (LDH) and lactate.34,590 This phenomenon is a result of the production of NADH by LDH: Serum (or plasma) is the most common specimen for ethanol analysis by ADH methods; this method also performs well with urine or oral fluid, although in some methods, whole blood may be used directly117 or a precipitation step may be required before analysis to avoid interference from hemoglobin.269 Results from these methods generally compare closely with those from gas chromatographic methods.117,291 For more information about these methods, see “Analysis of Volatile Alcohols” section, later. During the early part of the twentieth century, Dr. Erik M.P. Widmark, a Swedish physician, did much of the foundational research regarding alcohol pharmacokinetics in the human body. In addition, he developed an algebraic equation that allows one to estimate the amount of alcohol consumed by an individual or the associated blood alcohol concentration when the values of the other variables are given489,840: ρ (rho) = volume of distribution (L/kg) (0.68 for males, 0.55 for females) Ct = blood alcohol concentration (kg/L) β = rate of ethanol elimination (0.15 g/L/h) t = time since first drink (h) d = specific gravity of alcohol (0.8) Z = amount of ethanol alcohol per drink (L) (15 mL of ethanol in a standard drink)
Clinical Toxicology
Basic Information
Toxin
Antidote/Treatment14,68,185,251,722
Acetaminophen
N-Acetylcysteine
Aluminum, iron
Deferoxamine
Anticholinergic agents
Physostigmine
Arsenic
Dimercaprol; 2,3-dimercaptosuccinic acid; D-penicillamine
Barbiturates
Multiple-dose oral activated charcoal; alkaline diuresis (phenobarbital only)
Benzodiazepines
Flumazenil
Beta-blockers
Glucagon
Calcium channel blockers
Calcium; glucagon; high-dose insulin infusion
Carbamazepine
Multiple-dose oral activated charcoal; charcoal hemoperfusion
Carbon monoxide
Oxygen
Cyanide
Amyl nitrite, sodium nitrite, sodium thiosulfate; hydroxocobalamin
Digoxin
Anti-digoxin Fab fragments
Ethylene glycol, methanol
Fomepizole (4-methylpyrazol) or ethanol; hemodialysis
Isoniazid
Pyridoxine
Lead
Calcium disodium edetate; dimercaprol; 2,3-dimercaptosuccinic acid
Lithium
Hemodialysis
Mercury
Dimercaprol; 2,3-dimercaptosuccinic acid; D-penicillamine
Methanol
Fomepizole (4-methylpyrazol) or ethanol; hemodialysis; folate
Nitrites, nitrates
Methylene blue
Opioids
Naloxone
Organophosphate or carbamate
Atropine; pralidoxime (controversial for carbamates)
Salicylates
Bicarbonate; hemodialysis
Theophylline
Multiple-dose oral activated charcoal; hemodialysis
Tricyclic antidepressants
Bicarbonate; benzodiazepines
Clinical Considerations
Analytical Considerations
Clinical Evaluation
Secondary Survey
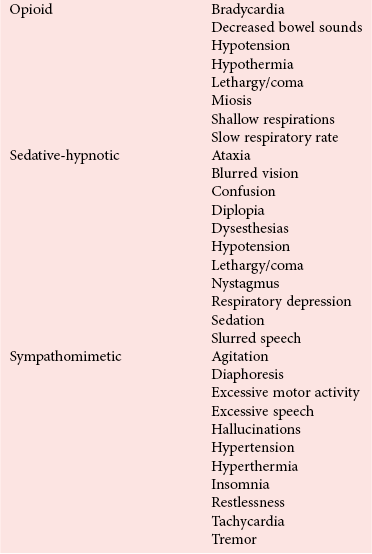
Toxic Syndromes
Cholinergic
Hyperthermic Syndromes
Screening Procedures for Detection of Drugs
Spot Tests
Ferric Chloride and Trinder Tests
Urine Fluorescence
Anion Gap
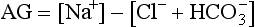
Electrocardiogram
Radiographic Studies
Osmol Gap
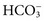 , glucose, and urea. Several empirical formulas based on measurement of these substances have been used to estimate the serum osmolality.276,539,606,617,662 In practice, one has not shown itself to be superior to the others, yet each equation demonstrates significant differences in the osmol gap reference interval.443 Therefore, reference intervals must be validated on appropriate patient populations. Two commonly used formulas (in conventional and SI units) are presented here:
, glucose, and urea. Several empirical formulas based on measurement of these substances have been used to estimate the serum osmolality.276,539,606,617,662 In practice, one has not shown itself to be superior to the others, yet each equation demonstrates significant differences in the osmol gap reference interval.443 Therefore, reference intervals must be validated on appropriate patient populations. Two commonly used formulas (in conventional and SI units) are presented here:
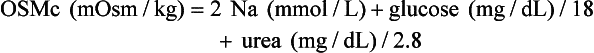
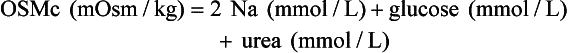
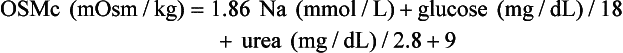
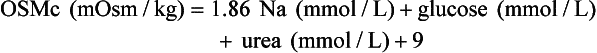
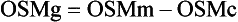
Immunoassay
Planar Chromatography
Gas Chromatography
High-Performance Liquid Chromatography
Point of Care (POC) Drug Testing
Pharmacology and Analysis of Specific Drugs and Toxic Agents
Agents That Cause Cellular Hypoxia
Carbon Monoxide
Toxic Effects
Analytical Methods
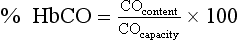
Cyanide
Toxic Effects
Analytical Methods
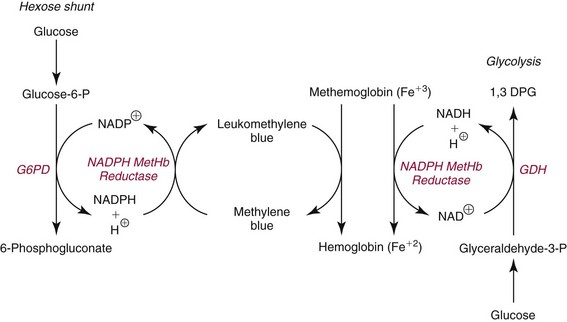
Methemoglobin-Forming Agents
Toxic Effects
Drugs
Chemical Agents
Amyl nitrite
Aniline
Benzocaine
Amyl nitrite
Chloroquine
Butyl nitrite
Dapsone
Chlorobenzene
Nitroglycerin
Naphthalene
Phenacetin
Nitrates
Phenazopyridine
Nitrites
Primaquine
Nitrophenol
Sulfonamides
Nitrous oxide
Analytical Methods
Alcohols of Toxicologic Interest
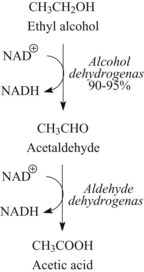
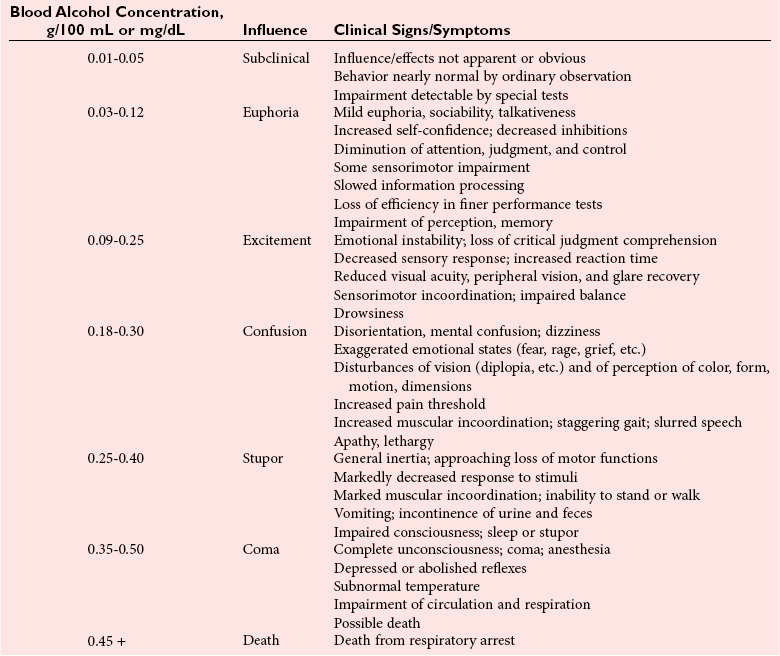
Ethanol
Methanol
Isopropanol and Acetone
Ethylene Glycol
Analysis of Ethanol
Serum/Plasma and Blood Ethanol
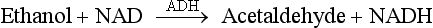
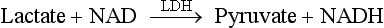
Estimation of Blood Alcohol
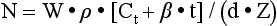
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree