Chapter 4 Serge Cremers; Jorge L. Sepulveda; Andrew Turk Questions During a diagnostic workup for hypertension and weight gain, a patient’s midnight salivary cortisol is found to be elevated (40 μg/dL; reference range, < 0.100 μg/dL) with a borderline decreased plasma adrenocorticotropic hormone (ACTH). The diagnosis of primary hyperadrenalism (Cushing’s syndrome) is considered. A. Measure urinary free cortisol. B. Measure a repeat salivary cortisol in the morning. C. Low-dose dexamethasone suppression test (DST). D. Measure plasma aldosterone. 1b. A diagnosis of Cushing’s syndrome is confirmed, and abdominal computed tomography (CT) reveals a 5-cm mass over the right adrenal gland. A high-dose DST is performed. Which one of the following is the most likely result of this testing? A. Greater than 50% reduction in serum cortisol. B. No suppression of serum cortisol. C. An elevation in serum cortisol. D. Suppression of serum metanephrines. A mother brings her 8-year-old son to the pediatrician for his annual checkup. She states that he seems to be struggling in school. Physical examination shows short stature and a palpable goiter. There is a family history of short stature and learning difficulties. The pediatrician performs thyroid function tests consisting of serum free thyroxine (FT4), free triiodothyronine (FT3), and thyroid-stimulating hormone (TSH). The results prompt additional workup. Ultimately, the boy is diagnosed with resistance to thyroid hormone (RTH). Use this scenario to answer the following four questions. 2a. Which one of the following choices most likely reflects this patient’s thyroid function studies? A. Normal TSH, decreased FT4, decreased FT3. B. Normal TSH, elevated FT4, elevated FT3. C. Elevated TSH, decreased FT4, decreased FT3. D. Elevated TSH, normal FT4, normal FT3. E. Elevated TSH, elevated FT4, elevated FT3. 2b. Given the physical examination findings (i.e., goiter) and the ultimate diagnosis of resistance to thyroid hormone (RTH), which one of the following mutations was most likely identified by genetic analysis? A. Inactivating mutation of the thyrotropin-releasing hormone receptor (TRHR) gene. B. Inactivating mutation of the thyroid stimulating hormone receptor (TSHR) gene. C. Inactivating mutation of the TSH β-chain gene. D. Inactivating mutation of the thyroxine-binding globulin (TBG) gene. E. Inactivating mutation of the thyroid hormone receptor (TR) β gene. 2c. Which one of the following statements is correct regarding the relative metabolic activity of thyroxine (T4), triiodothyronine (T3), and reverse triiodothyronine (rT3)? A. T4 is more metabolically active than T3, which is more metabolically active than rT3. B. T4 is more metabolically active than rT3, which is more metabolically active than T3. C. T3 and rT3 are approximately equally metabolically active. These hormones are more metabolically active than T4. D. T3 is more metabolically active than T4, which is more metabolically active than rT3. E. T3 is more metabolically active than rT3, which is more metabolically active than T4. 2d. Certain organs show greater impairment than others in the setting of hypothyroidism. Which one of the following organs is likely to show the least functional impairment in a hypothyroid patient? B. Kidney. C. Spleen. D. Skeletal muscle. E. Heart. During his annual checkup, a 34 year-old white man complains of a chronically low libido to his primary care physician. The patient’s history includes acute leukemia during his early childhood, which was treated with chemotherapy. Physical examination reveals tall stature and gynecomastia. Laboratory workup reveals a serum total testosterone level of 150 ng/dL (reference range, 270 to 1070 ng/dL). Use this scenario to answer the following four questions. 3a. Careful physical examination of this patient is most likely to show which one of the following features? A. Decreased fat deposition in the hips, buttocks, and thighs. B. Increased fat deposition in the face, abdomen, and waist. C. Decreased facial and body hair. D. Decreased hair on the temporal aspect of the scalp. E. A diamond-shaped escutcheon (pubic hair pattern), rather than a triangular escutcheon. 3b. Serum total testosterone levels are most likely to be normal in patients with which one of the following genetic conditions? B. XYY syndrome. C. Kallmann’s syndrome. D. Loss-of-function mutations in the gene encoding GPR54. E. CHARGE syndrome. 3c. Which one of the following statements best explains this patient’s tall stature? B. Estrogens ultimately terminate growth by causing closure of the epiphyses of the long bones. C. Androgens have a net anabolic effect in terms of protein metabolism (i.e., androgens increase protein synthesis and decrease protein degradation). D. Androgens increase the deposition of calcium salts and the rate of bone growth. E. Boys who undergo precocious puberty (i.e., onset of puberty before age 9 years) typically have tall stature as adults. 3d. Regarding demographic and physiologic parameters that affect normal serum testosterone levels in adult men, which one of the following statements is true? B. The quantitative relationship between serum total testosterone and serum free testosterone depends primarily on the serum concentration of albumin. C. Serum testosterone levels are inversely proportional to body mass index (BMI). D. Serum testosterone levels have no quantitative relationship with central obesity (i.e., waist/hip ratio). E. Serum total testosterone levels are higher in young white men compared with young black men. A 35-year-old woman was brought to the emergency department after developing psychosis. Laboratory studies performed in the emergency department revealed hyperglycemia and hypokalemia. Further studies performed during her subsequent hospital course revealed increased 24-hour urine free cortisol (5796 nmol; reference range, 55 to 193 nmol), consistent with Cushing’s syndrome. Imaging studies revealed a left adrenal mass. Use this scenario to answer the following four questions. 4a. Which one of the following statements is true? B. The increased urine free cortisol indicates a functional adrenal cortical neoplasm and effectively rules out the possibility of a malignant neoplasm. C. Cushing’s syndrome caused by ectopic production of ACTH occurs more frequently than ACTH-independent Cushing’s syndrome. D. The patient’s psychosis is probably related to her adrenal mass. E. The patient’s hypokalemia is probably due to increased intracellular potassium uptake caused by high serum cortisol levels. 4b. Given the patient’s elevated urine free cortisol, additional laboratory testing would likely show increased levels of other glucocorticoids and glucocorticoid precursors. Which one of the following molecules is a mineralocorticoid precursor, rather than a glucocorticoid, glucocorticoid precursor, or biosynthetic precursor of cortisol? B. Progesterone. C. 11-Deoxycortisol. D. Corticosterone. E. Cortisone. 4c. Which one of the following statements explains hyperglycemia within the context of Cushing’s syndrome? B. Cortisol decreases hepatic gluconeogenesis by accelerating protein degradation, thereby increasing the amounts of amino acid substrates for gluconeogenesis. C. Cortisol directly activates key hepatic gluconeogenic enzymes and increases the activity of hepatic glycogen synthetase. D. Cortisol increases the uptake and phosphorylation of glucose by peripheral tissues. 4d. Which one of the following statements regarding hormones produced by adrenal tumors is true? B. Virilizing tumors of the adrenal gland occur rarely, cause clinical changes in women but not in men, and are associated with low levels of dehydroepiandrosterone sulfate (DHEA-S) in serum and 17-ketosteroids in urine. C. Pheochromocytomas are associated with multiple endocrine neoplasia syndrome, types IIa (MEN IIa) and IIb (MEN IIb), von Hippel–Lindau syndrome, familial paraganglioma syndrome, and neurofibromatosis I. D. Neuroblastoma is a well-differentiated tumor of childhood that rarely produces catecholamines. A 36-year-old man sought help from a reproductive endocrinologist for a workup of infertility. Physical examination showed normal male genitalia. Hormonal analysis revealed markedly increased levels of serum 17-hydroxyprogesterone and reduced levels of serum 11-deoxycortisol. Circulating levels of other hormones were with normal limits. Semen analysis showed a reduced percentage of viable sperm and a reduced percentage of morphologically normal sperm. Sperm concentration and percentage of motile sperm were within normal limits. Use this scenario to answer the following three questions. 5a. Which one of the following is the most likely diagnosis? B. 21-Hydroxylase deficiency. C. 11-β-Hydroxylase deficiency. D. 3-β-Hydroxylase deficiency. E. Androgen insensitivity syndrome (AIS). 5b. Thorough evaluation of this patient would most likely reveal increased levels of which one of the following hormones? B. Aldosterone. C. 18-Hydroxycorticosterone. D. Corticosterone. E. Cortisol. 5c. Which one of the following features would most likely characterize a female patient with this condition? A. Decreased serum levels of pregnenolone. B. Increased serum levels of 11-deoxycorticosterone. C. Decreased serum levels of dehydroepiandrosterone. D. Decreased serum levels of progesterone. E. An abnormal physical examination. A 38-year-old woman with polycystic ovarian syndrome (PCOS) consulted with a reproductive endocrinologist regarding in vitro fertilization (IVF). After a comprehensive review of the patient’s history and a thorough physical examination, the physician ordered a battery of laboratory tests. Use this scenario to answer the next four questions. 6a. Which one of the following hormones is most likely to be within normal limits in this patient’s serum? B. Androstenedione. C. Luteinizing hormone (LH). D. Follicle-stimulating hormone (FSH). E. Insulin. 6b. Additional workup revealed abnormalities of anti-Müllerian hormone (AMH). Which one of the following options most accurately describes abnormalities associated with AMH in patients with PCOS? A. AMH levels are elevated in serum but not in ovarian follicular fluid. B. AMH levels are elevated in ovarian follicular fluid but not in serum. C. AMH levels are elevated in serum and in ovarian follicular fluid. D. AMH levels are not elevated in serum or ovarian follicular fluid. E. In patients with PCOS, the abnormality of AMH is qualitative rather than quantitative. 6c. Which one of the following is likely to show abnormalities of AMH similar to those associated with PCOS? A. Pelvic inflammatory disease (PID). B. Salpingitis isthmica nodosa (SIN). C. Endometriosis. D. Pelvic adhesions from prior surgery. E. Granulosa cell tumors. 6d. Which one of the following explanations has been proposed to account for the chronic anovulatory state of patients with PCOS? B. Low serum LH levels prevent primordial follicles from initiating the process of maturation. C. AMH activity prevents primordial follicles from initiating the process of maturation. D. AMH stimulates aromatase activity within granulosa cells, thereby reducing production of estradiol. A 45-year-old woman underwent surgical resection of a granulosa cell tumor, a representative image of which is shown in Figure 4-1. Use this scenario to answer the following three questions. 7a. Which one of the following statements is correct in terms of the hormones secreted by these tumors? B. Granulosa cell tumors secrete peptide hormones; secretion of steroid hormones by these tumors has not been reported. C. Granulosa cell tumors secrete steroid hormones (including estrogen, testosterone, progestins, and inhibin), as well as peptide hormones (including AMH). D. Granulosa cell tumors secrete steroid hormones (including estrogen, testosterone, and progestins), as well as peptide hormones (including AMH and inhibin). E. Granulosa cell tumors are generally poorly differentiated neoplasms, and production of functional hormones by these lesions is exceedingly rare. 7b. Granulosa cell tumors are most likely to cause elevation of which one of the following tumor markers? B. β-Human chorionic gonadotropin (β-hCG). C. Carcinoembryonic antigen (CEA). D. Lactate dehydrogenase (LDH). E. CA-125. 7c. Which one of the following options most accurately conveys the clinical features of granulosa cell tumors? B. Granulosa cell tumors frequently behave aggressively in children and young adults but generally follow a benign course in middle-aged and postmenopausal patients. C. Granulosa cell tumors frequently behave aggressively in younger and older patients alike. D. Granulosa cell tumors generally follow a benign course in younger and older patients alike. E. Granulosa cell tumors generally follow a benign course in middle-aged and postmenopausal patients; these tumors have not been described in children and young adults. A 35-year-old man is found on routine examination to have a blood pressure of 165/110 mm Hg without any other symptoms. Laboratory values are shown in Table 4-1. Urine electrolyte measurements resulted in sodium excretion of 216 mmol/24 hours (reference range, 40 to 220 mmol/24 hr) and potassium 135 mmol/24 hours (reference range, 25 to 125 mmol/24 hr). Use this scenario to answer the following three questions. 8a. Which one of the following disorders best explains these results? B. Graves’ disease. C. Conn’s syndrome. D. Mild congenital adrenal hyperplasia. E. Panhypopituitarism. 8b. Which one of the following laboratory test results is most consistent with the diagnosis? A. Elevated plasma renin activity (PRA) and decreased aldosterone. B. Suppressed PRA and elevated aldosterone. C. Elevated 17-hydroxyprogesterone and low aldosterone. D. Elevated FT4 and suppressed TSH. E. Low TSH and low FT4, 8c. Which one of the following confirmatory test results is most consistent with the patient’s diagnosis? A. Continued suppression of aldosterone after stimulation with adrenocorticotropic hormone. B. Failure to increase TSH after stimulation with thyroid-releasing hormone. C. Maintained aldosterone elevation after infusion of 2000 mL of isotonic saline. D. Elevated 17-hydroxyprogesterone after ACTH stimulation. E. Failure to increase cortisol after insulin-induced hypoglycemia. 9. A 17-year-old woman was referred to the endocrinologist for primary amenorrhea. On physical examination, she had minimal axillary and pubic hair and Tanner stage II breast development. Which one of the following laboratory results would have the highest diagnostic value? B. DHEA-S. C. Androstenedione. D. Progesterone. E. β-hCG. 10. A 45-year-old woman presents with lack of menstruation for 7 months, proptosis, headaches, and personality changes. A pregnancy test result was repeatedly negative. Laboratory evaluation was remarkable for a prolactin level of 65 ng/mL (reference range, 3.8 to 23.2 ng/mL). Magnetic resonance imaging (MRI) of the head revealed a 3-cm mass in the base of the skull. Which one of the following most likely explains the patient’s symptoms and elevation in prolactin levels? A. Large prolactinoma and hook effect. B. Non–prolactin-secreting tumor and macroprolactin. C. Stress-associated hyperprolactinemia. D. Graves’ disease. E. Unreported opiate use. 11. A 20-year-old woman was diagnosed with primary amenorrhea. On physical examination, she had a normal female habitus and voice, normal breast development, and absent axillary and pubic hair. Family history was unremarkable. Pelvic ultrasound showed absence of a uterus and ovaries and the presence of undescended testes. Her karyotype was 46,XY. Which one of the following laboratory results is most consistent with the patient’s diagnosis? B. Decreased LH and FSH. C. Low estradiol. D. Elevated 17-hydroxyprogesterone. E. Mutated androgen receptor. 12. As part of her bone health checkup, C-terminal telopeptide (CTX) and N-terminal propeptide of type I collagen (PINP) are measured by automated chemiluminescent (CLIA) and radioimmunoassay (RIA) in a morning serum sample of a 62-year-old woman with postmenopausal osteoporosis (bone mineral density T-score, − 2.8). CTX was 0.738 ng/mL (reference range, 0.115 to 0.748 ng/mL) and PINP was 60 ng/mL (reference range, 19 to 83 ng/mL). One month later, these bone markers are remeasured, this time in a morning fasting sample tested by a reference laboratory rather than your hospital laboratory. CTX was 1.032 ng/mL and PINP was 62 ng/mL. The patient has been taking vitamin D supplements for more than 2 years. Which one of the following is the most likely explanation for why CTX is higher while PINP is relatively unchanged? B. CTX reflects significant improvement in osteoporosis, whereas PINP is falsely elevated because of the influence of food intake and circadian variability on PINP levels. C. The difference between the two CTX levels is within the expected analytical variability. D. Influence of food intake on CTX levels and circadian variability in CTX levels. E. Intralaboratory variability in CTX and PINP results. 13. A 32-year-old woman is admitted to the hospital with paresthesia and seizures. A total calcium level, determined in plasma ethylenediaminetetraacetic acid (EDTA) by a colorimetric method using arsenazo III, is 8.5 mg/dL (reference range, 8.6 to 10.1 mg/dL). Albumin is 3.8 mg/dL (reference range, 3.5 to 5.2 g/dL). Which one of the following would be the most appropriate next step in the management of this patient? A. The patient should be treated for hypocalcemia. B. The sample should be sent to a reference laboratory to determine calcium by atomic absorption spectrometry (AAS). C. The measurement should be repeated using serum. D. The patient should not be treated for hypocalcemia E. Ionized calcium should be measured in the EDTA sample. A 35-year-old pregnant woman with a history of hyperthyroidism (Graves’ disease) was assessed for the presence of TSH receptor (TSHR) antibodies (TRAb) during a third-trimester prenatal evaluation. Her thyroid-binding inhibitory immunoglobulin (TBII) levels measured by quantitative chemiluminescence immunoassay were 15.0 U/mL (reference range, < 1.75 U/mL), and her thyroid peroxidase (TPO) antibody levels were 150 U/mL (reference range, < 9 U/mL). Her FT4 levels were 1.45 ng/dL (reference range, 0.70 to 1.24 ng/dL), and her TSH levels were less than 0.01 mIU/L (reference range, 0.32 to 4.04 mIU/L). Use this scenario to answer the following three questions. 14a. Which one of the following statements is most consistent with these findings? A. The patient has active Graves’ disease. B. This patient has gestational thyrotoxicosis. C. The presence of anti-TPO antibodies rules out Graves’ disease. D. This patient is undergoing effective anti-thyroid drug treatment. E. This patient has Hashimoto’s thyroiditis and not Graves’ disease. 14b. Which one of the following statements regarding the risk for neonatal thyroid disease in this case is correct? A. The risk for neonatal thyroid disease is the same as in the general population. B. The fetus is at risk for neonatal hyperthyroidism due to transplacental transport of TRAb. C. The fetus is at risk for neonatal hypothyroidism due to transplacental transport of thyroid-blocking antibodies. D. The fetus is at risk for hypothyroidism due to overtreatment of the mother’s hyperthyroidism with antithyroid medications. E. The fetus is at risk for neonatal hypothyroidism due to Hashimoto’s thyroiditis. 14c. The mother was treated with 100 mg of propylthiouracil (PTU) four times per day, resulting in normalization of FT4 and TSH levels and delivery of a normal infant. PTU treatment was discontinued postpartum. Six months postpartum, the patient complained of facial puffiness, depression, lack of energy, and insomnia. Evaluation of thyroid function showed FT4 levels of 0.20 ng/dL (reference range, 0.70 to 1.24 ng/dL), TSH levels of 290 mIU/L (reference range, 0.4 to 5.0 mIU/L), thyroid-binding inhibitory immunoglobulin levels of 10.75 U/mL (reference range, < 1.75 U/mL), and anti−thyroid peroxidase antibody levels of 15 U/mL (reference range, < 9 U/mL). A cell culture assay for thyroid-stimulating antibodies (TSAb) in the patient’s serum showed levels of 5% (reference range, < 11%). Which one of the following statements is most consistent with these findings? A. The patient has postpartum depression but is euthyroid. B. The patient has atrophic hypothyroidism due to overtreatment with PTU. C. The patient now has hypothyroidism due to autoimmune thyroiditis. D. The patient developed anti-TSH antibodies. E. This patient now has thyroid-blocking antibody-induced hypothyroidism. A 67-year-old man with multiple health problems sought medical attention for difficulty sleeping. Physical examination revealed markedly reduced vibratory sensation in his legs. Laboratory workup showed low TSH (< 0.03 mIU/L; reference range, 0.4 to 5.0 mIU/L), high free thyroxine (T4) (2.42 ng/dL; reference range, 0.70 to 1.24 ng/dL), negative anti−thyroid peroxidase (anti-TPO) antibodies, normal white blood cell count (4.1 × 109/L; reference range, 3.5 to 9.1 × 109/L), and normal erythrocyte sedimentation rate (13 mm/hr; reference range, 1 to 15 mm/hr). Nuclear medicine studies showed low radionuclide uptake. Collectively, these findings indicate thyrotoxicosis without underlying hyperthyroidism. He underwent total thyroidectomy, a representative image of which is shown in Figure 4-2. Use this scenario to answer the following three questions. 15a. Which one of the following drugs did this patient most likely receive before the development of his symptoms? B. Interleukin-2. C. Amiodarone. D. Minocycline. 15b. Which one of the following forms of thyroiditis would most likely be associated with the clinical scenario and laboratory findings described above (i.e., thyrotoxicosis without underlying hyperthyroidism)? B. Subacute thyroiditis. C. Hashimoto’s thyroiditis. D. Riedel’s thyroiditis. 15c. Which one of the following hyperplastic or neoplastic conditions (or sequelae thereof) would most likely be associated with the clinical scenario and laboratory findings described above? A. Diffuse follicular epithelial hyperplasia. B. Follicular adenoma. C. Infarcted adenoma. D. Medullary thyroid carcinoma. 16. 25(OH)-Vitamin D is measured in the serum of a 6-month-old boy using three different techniques: DiaSorin Liaison TOTAL (CLIA), Immunodiagnostics-iSYS (CLIA), and liquid chromatography/tandem mass spectrometry (LC-MS/MS). Using the latter, the concentration of 25(OH)-vitamin D2 was below the level of detection of 1 ng/mL and the concentration of 25(OH)-vitamin D3 was 35 ng/mL. The 25(OH)-vitamin D serum concentrations measured with the DiaSorin Liaison TOTAL and the Immunodiagnostics-iSYS instruments were 25 and 26 ng/mL, respectively. Which one of the following reasons best explains the discrepancy between the results obtained by LC-MS/MS and those obtained by the automated immunochemistry analyzers? B. The DiaSorin Liaison TOTAL and the Immunodiagnostics-iSYS CLIA methods both detect 25(OH)-vitamin D2 and 25(OH)-vitamin D3. C. The DiaSorin Liaison TOTAL and the Immunodiagnostics-iSYS CLIA methods do not detect 25(OH)-vitamin D2. D. The DiaSorin Liaison TOTAL and the Immunodiagnostics-iSYS CLIA methods detect both the 25(OH)-vitamin D3 C3-epimer and 25(OH)-vitamin D3. E. Most mass spectrometry−based assays do measure the 25(OH)-vitamin D3 C3-epimer but cannot distinguish it from 25(OH)-vitamin D3. 17. Which one of the following best describes serum hCG levels in pregnancy? B. Molar pregnancies are associated with lower than expected serum hCG levels. C. In a normal pregnancy, maximum serum hCG levels are reached at or near the end of the first trimester. D. In a normal pregnancy, serum hCG levels return to nonpregnant levels within 2 to 3 days after a normal delivery. E. In a normal pregnancy, serum hCG levels are at their highest in the third trimester. 18. An 18-year-old man presents with renal colic. A calcium oxalate kidney stone is passed several days later. His serum levels are shown in Table 4-2 and his 24-hour urine levels are shown in Table 4-3. Which one of the following answers contains the best explanation for this patient’s hypercalciuric nephrolithiasis and the biochemical test that should be done to support this diagnosis? A. Decreased CYP24A1 activity. Measure serum 24,25(OH)-vitamin D. B. Hypoparathyroidism. Measure parathyroid hormone (PTH) with a third-generation assay. C. Cancer. Measure PTH-related peptide. D. Chronic kidney disease. Measure neutrophil gelatinase−associated lipocalin (NGAL). E. Familial hypocalciuric hypercalcemia. Measure serum phosphate. 19. An otherwise healthy 28-year-old woman has incompletely understood symptoms suggestive of hypomagnesemia. However, a total serum magnesium (Mg) level determined colorimetrically was normal. Now, an ionized serum Mg level is requested to acquire additional information. Which one of the following reasons would be the best explanation of a falsely low ionized Mg level in this patient? A. There is a weak relationship between ionized and total Mg levels in serum. B. Interference by cotinine in the serum from cigarette smoking by the patient. C. Interference by thiocyanate in the serum from cigarette smoking by the patient. D. Vitamin C intake by the patient. E. The presence of hypoalbuminemia. 20. At 3 to 5 weeks of gestation, which one of the following is the predominant form of hCG that is detected in serum? B. Intact hCG. C. Free β-hCG. D. Nicked hCG. E. Pituitary hCG. 21. Several bone turnover markers are measured in the serum and urine of a cancer patient. Serum levels (nonfasting, collected at 2 pm) are as follows: ■ Bone alkaline phosphatase = 18.7 U/L (reference range, 11.5 to 29.6 U/L) Levels in a 24-hour urine sample are as follows: Given these findings, which one of the following types of cancer is most likely present in this patient? A. Prostate cancer with bone metastasis. B. Breast cancer with bone metastasis. C. Multiple myeloma with osteolytic lesions. D. Renal carcinoma with bone metastasis. E. Bone metastases from non−small cell lung carcinoma. 22. A 57-year-old white man has been complaining for 2 years about pain in his right humerus. The skin overlying the painful bone feels warm. The bone itself feels smooth. Otherwise, the patient is healthy and does not show any skeletal deformations. To his knowledge there is no family history of bone disease. Serum levels are shown in Table 4-4. X-rays show focal osteolysis with coarsening of the trabecular pattern, bone expansion, and cortical thickening. A technetium-99 m (99mTc) diphosphonate bone scan shows increased uptake in the upper part of the right humerus. Which one of the following is the most likely diagnosis? B. Multiple myeloma. C. Paget’s disease of bone. D. Male osteoporosis. E. Van Buchem’s disease. 23. A 46-year-old woman in good health was seeing her internist as part of a routine physical examination. Because she reported 6 months of amenorrhea, serum and urine hCG levels were measured to rule out pregnancy. The serum hCG level was 10 IU/L (reference range, 5 to 20 IU/L), and a positive urine hCG result was obtained. Serum hCG was measured in another sample 7 days later and was 12 IU/L. When hCG was measured with a different immunochemical method, the serum hCG result was 11 IU/L. The patient’s serum FSH level was 130 IU/L (reference range, ≥ 45 IU/L). Which one of the following is the most probable cause of the detectable hCG level in this patient? A. Positive interference from human anti-animal antibodies. B. Normal intrauterine pregnancy. C. Ectopic pregnancy. D. Synthesis and release of hCG from the pituitary gland. E. Presence of a hydatidiform mole. 24. Intact PTH is measured in a plasma sample from a 42-year-old man using an RIA (Scantibodies, Santee, CA). The concentration is 75 pg/mL (reference range, 14 to 66 pg/mL). Whole PTH is also measured in the same plasma sample using an RIA (Scantibodies). The concentration is 37 pg/mL (reference range, 6 to 32 pg/mL). The patient’s height, body weight, and serum creatinine are 186 cm, 85 kg, and 0.8 mg/dL (reference range, 0.6 to 1.2 mg/dL), respectively. Serum calcium and albumin are 10.3 mg/dL (reference range, 9 to 10.5 mg/dL) and 4.5 mg/dL (reference range, 3.5 to 5.5 g/dL), respectively. Which one of the following statements best explains these results? A. The patient has primary hyperparathyroidism. B. The patient has hyperparathyroidism secondary to kidney disease. C. The patient has a parathyroid carcinoma. D. Intact PTH assays always give a higher result than the whole PTH assays. E. RIA methods are unreliable for measuring PTH levels. 25. A 59-year-old man with a history of type 2 diabetes and hyperlipidemia is referred to your hospital for evaluation of recently discovered hypercalcemia. His laboratory results from blood samples are shown in Table 4-5. In addition, his liver function tests, thyroid function tests, urine metanephrines, serum angiotensin-converting enzyme (ACE) activity, and serum and urine protein and electrophoresis analyses are all normal. The intact PTH level was measured by an immunochemiluminescent assay (ICMA). Ultrasonography of the neck did not reveal any abnormalities of the thyroid or parathyroid glands, but a 99mTc-sestamibi scan suggests the presence of a parathyroid adenoma. Bone mineral densities by dual x-ray absorptiometry (DXA) scanning of the lumbar spine, femoral neck, and radius are − 1.7, − 1.3, and − 1.7, respectively, indicating osteopenia. Which one of the following is the best explanation for the unusual combination of results for calcium and intact PTH in this case? A. Serum samples were kept on ice during transport. B. Vitamin D deficiency. C. Circulating variant forms of PTH. D. Hypermagnesemia. E. The hook effect. 26. An 18-year-old obese man with short stature, a round face, subcutaneous ossifications, and brachydactyly undergoes a biochemical workup by an endocrinologist. His laboratory results from blood samples under fasting conditions are shown in Table 4-6. His laboratory results from a 24-hour urine collection are shown in Table 4-7. Which one of the following is the most likely diagnosis in this patient? A. Primary hyperparathyroidism. B. Secondary hyperparathyroidism. C. Hypoparathyroidism. D. Pseudohypoparathyroidism. E. Pseudopseudohypoparathyroidism. 27. In which one of the following situations is 1,25(OH)-vitamin D expected to be elevated? A. Secondary hyperparathyroidism. B. Hypoparathyroidism. C. Postmenopausal osteoporosis. D. Sarcoidosis. E. Hypercalcemia of malignancy. 28. A 52-year-old morbidly obese man underwent bariatric surgery in 2005. Three years later, he was seen by an ophthalmologist for progressive loss of vision and night blindness. Blood work performed as part of a routine checkup showed mild hypercalcemia and mild hyperparathyroidism. Measuring which one of the following is the most appropriate and most specific test to account for his impaired night vision? B. 25(OH)-vitamin D. C. 1,25(OH)-vitamin D. D. Total protein. E. Albumin. 29. You are asked to measure the zinc (Zn) level in a specimen from a 23-year-old woman to confirm suspected Zn deficiency. You measure Zn in your laboratory by flame atomic absorption spectroscopy (AAS). Which one of the following settings could lead to higher Zn levels? B. The use of prednisone by the patient. C. Obtaining a blood sample in the evening. D. Measuring Zn in serum rather than plasma. E. Recent food intake. 1b. A. Greater than 50% reduction in serum cortisol. Major points of discussion ■ There are multiple etiologies for Cushing’s syndrome, but all result in excess production of cortisol by the adrenal glands or hyperadrenalism. ■ Laboratory testing reveals increased levels of serum cortisol, urinary free cortisol, lack of suppression of cortisol production after administration of low-dose dexamethasone overnight, and hyperglycemia. ■ Cushing’s syndrome may result from multiple etiologies, including iatrogenic administration, ectopic production of corticotropin-releasing hormone (CRH) or ACTH, adrenal carcinoma or adenoma, and an ACTH-producing pituitary adenoma or Cushing’s disease. The treatment and prognosis are dependent on the etiology. ■ Screening tests for hyperadrenalism include 24-hour urinary free cortisol, overnight low-dose DST, and a midnight salivary cortisol. ■ The low-dose DST is useful in testing to determine which portion of the hypothalamic-pituitary-adrenal axis is impaired in hyperadrenalism. The normal response to 2 days of low-dose dexamethasone is suppression of serum cortisol levels. Lack of suppression suggests primary hyperadrenalism, or Cushing’s syndrome. ■ In cases of hyperadrenalism with borderline depressions in ACTH, the best diagnostic test is the low-dose DST. ■ A high-dose overnight DST is used to help differentiate between the different causes of Cushing’s syndrome. Suppression of cortisol production with increased ACTH suggests Cushing’s disease or ectopic production of ACTH. Demonstration of a pituitary mass on MRI or CT is diagnostic of Cushing’s disease. If a pituitary mass is not present, CT of the chest and abdomen may demonstrate an ACTH-producing neoplasm. Failure to suppress after high-dose dexamethasone and increased ACTH levels suggests ectopic ACTH production, and CT of the chest and abdomen is warranted. Failure to suppress after high-dose dexamethasone and low ACTH levels suggests the hyperadrenalism may be of adrenal origin. CT of the adrenals may demonstrate an adrenal tumor. 2a. A. Normal TSH, decreased FT4, decreased FT3. 2b. A. Inactivating mutation of the thyrotropin-releasing hormone receptor (TRHR) gene. 2c. A. T4 is more metabolically active than T3, which is more metabolically active than rT3. 2d. A. Liver. Major points of discussion ■ Among the various etiologies of congenital hypothyroidism, errors that decrease the quantity or functionality of TSH (including errors that are “upstream” of TSH) and errors that affect thyroid hormone transport do not cause goiter. In general, this applies to categories (1), (2), and (4), as outlined above. ■ Congenital hypothyroidism caused by errors in thyroid hormone synthesis and action (e.g., RTH) generally result in goiter formation, owing to a compensatory increase in TSH. This applies to categories (3) and (5) as outlined above. ■ RTH is a rare syndrome of reduced end-organ responsiveness to thyroid hormone. Because of a compensatory feedback mechanism, patients with RTH have elevated T4, T3, and serum rT3. ■ T4 is three to four times more metabolically active than T3. T4 is a precursor hormone that can be transformed into T3 by type I or type II iodothyronine deiodinase. Type III iodothyronine deiodinase inactivates T4 and T3 by producing rT3. ■ Liver, kidney, and skeletal muscle are among the most thyroid-responsive tissues. Spleen, testes, and (adult) brain are relatively thyroid unresponsive.28,37 3a. A. Decreased fat deposition in the hips, buttocks, and thighs. 3b. A. Klinefelter’s syndrome. 3c. A. Androgens stimulate increased secretion of insulin-like growth factor 1 (IGF-1), which in turn causes growth. 3d. A. Serum total testosterone levels decrease with age, whereas serum free testosterone levels remain relatively constant with age. Major points of discussion ■ In central hypogonadism, the hypothalamus and/or pituitary does not function properly. Some causes of central hypogonadism include genetic and developmental disorders (e.g., Kallmann’s syndrome), central nervous system tumors, nutritional deficiencies, infection, medications (e.g., steroids, opiates), radiation, and trauma. ■ Men with hypogonadism with onset before puberty typically demonstrate tall stature, decreased facial and body hair, gynecomastia, a female or triangular escutcheon (rather than a male or diamond-shaped escutcheon), a female fat distribution pattern (increased fat deposition in the hips/buttocks/thighs, decreased fat deposition in the face/abdomen/waist), and lack of temporal hair recession. These men are also at increased risk for osteoporosis later in life. ■ Androgens affect bone growth during puberty by various mechanisms. Androgens increase the amplitude of the “spikes” in the pulsatile secretion of growth hormone. This effect increases secretion of IGF-1, which in turn causes growth. Androgens also facilitate bone growth by increasing protein synthesis, decreasing protein degradation, and increasing the deposition of calcium salts. These effects account for the growth changes seen in boys during puberty. Estrogens, which are derived from androgens, ultimately terminate growth by causing closure of the epiphyses of the long bones. ■ In healthy adult men, serum total testosterone and serum free testosterone levels decrease with age. The quantitative relationship between serum total testosterone and serum free testosterone levels depends primarily on the serum concentration of sex hormone–binding globulin. Cross-sectional epidemiologic studies have shown an inverse relationship between BMI and serum testosterone levels, as well as an inverse relationship between waist/hip ratio and serum testosterone levels.26,60 4a. A. The combination of hypokalemia, elevated urine free cortisol, and an adrenal mass indicates a neoplasm of adrenal cortical origin and rules out the possibility of a pheochromocytoma. 4b. A. Pregnenolone. 4c. A. Cortisol decreases hepatic gluconeogenesis by inhibiting protein synthesis, thereby increasing the amounts of amino acid substrates for gluconeogenesis. 4d. A. Primary hyperaldosteronism (Conn’s syndrome) is characterized by hyperkalemia and hypotension (among other features) and is almost always due to an adenoma of the adrenal cortex. Major points of discussion ■ Cortisol facilitates the net conversion of protein to glycogen. Cortisol simultaneously increases glycogen stores in the liver (by increasing the activity of glycogen synthetase), as well as production of glucose by the liver. Cortisol increases hepatic glucose production by facilitating gluconeogenesis (i.e., by inhibiting protein synthesis, accelerating protein degradation, and directly activating gluconeogenic enzymes). Cortisol also decreases the uptake and phosphorylation (i.e., catabolism) of glucose by peripheral tissues. ■ Steroid hormones produced by the adrenal gland (e.g., cortisol) are ultimately derived from cholesterol. Cholesterol is converted into pregnenolone, which is in turn converted into progesterone. Progesterone, 17-hydroxyprogesterone, and 11-deoxycortisol are considered glucocorticoid precursors. 11-Deoxycortisol is converted into cortisol, which is in turn converted into cortisone; cortisol and cortisone are considered active glucocorticoids. ■ Pheochromocytomas can mimic functional adrenal cortical tumors in terms of their clinical features (e.g., by causing the clinical scenario described in this case). Functional tumors are not always benign; hormone-producing adrenal cortical carcinomas and catecholamine-producing malignant pheochromocytomas are well-documented entities. ACTH-independent Cushing’s syndrome caused by adrenal tumors is approximately twice as common as ACTH-dependent Cushing’s syndrome caused by ectopic production of ACTH. ■ Various other tumors arising in the adrenal gland produce hormones. Primary hyperaldosteronism (Conn’s syndrome) is almost always due to an adenoma of the adrenal cortex. Virilizing tumors of the adrenal occur rarely, cause clinical changes in women but not in men, and are associated with high levels of DHEA-S in serum and 17-ketosteroids in urine. Neuroblastoma is a poorly differentiated neoplasm derived from neural crest cells, usually occurring in infants and small children. From 50% to 80% arise in the adrenal gland, and more than 90% produce catecholamines.35,36,56 5a. A. Addison’s disease. 5b. A. 17-Hydroxypregnenolone. 5c. A. Decreased serum levels of pregnenolone. Major points of discussion ■ The adrenal cortex derives from intermediate mesoderm and produces steroid hormones. The cortex consists of three layers (from outermost to innermost: the zona glomerulosa, zona fasciculata, and zona reticularis). The renin-angiotensin system modulates the function of the zona glomerulosa, whereas the hypothalamic-pituitary axis regulates the zona fasciculata and zona reticularis. ■ The adrenal medulla derives from neural crest (ectoderm) and produces peptide hormones (i.e., catecholamines, enkephalins, chromogranins, and others). The splanchnic nerve regulates the function of the medulla. ■ Adrenal cortical cells produce three types of steroid hormones: mineralocorticoids, glucocorticoids, and sex hormones. Mineralocorticoids, synthesized primarily in the zona glomerulosa, mediate renal tubular reabsorption of sodium and water. Glucocorticoids, produced primarily in the zona fasciculata, facilitate the net conversion of protein to glycogen (among myriad other functions). Sex hormones (androgens and estrogens) are the major products of the zona reticularis. ■ Mineralocorticoids include aldosterone, 18-hydroxycorticosterone, corticosterone, and 11-deoxycorticosterone. Glucocorticoids (e.g., cortisol, 11-deoxycortisol, 17-hydroxyprogesterone, and 17-hydroxypregnenolone) consist of 21 carbons and are hydroxylated at position 17. Androgens (e.g., dehydroepiandrosterone, androstenedione, androstenediol, and testosterone) contain 18 carbons, whereas estrogens (e.g., estrone and estradiol) contain 17 carbons. Estrogens also possess aromatic A rings. ■ Enzymatic deficiencies that affect the biosynthesis of cortisol are collectively known as congenital adrenal hyperplasia. These disorders include 21-hydroxylase deficiency, 11-β-hydroxylase deficiency, 3-β-hydroxylase deficiency, 17-β-hydroxylase deficiency, and lipoid hyperplasia. The manifestations of these disorders are heterogeneous and depend on the severity of the patient’s enzymatic defect. ■ 21-Hydroxylase converts progesterone into 11-deoxycorticosterone (a precursor of aldosterone) and 17-hydroxyprogesterone into 11-deoxycortisol (a precursor of cortisol). Patients with 21-hydroxylase deficiency consequently have relatively low levels of mineralocorticoids (which are downstream of progesterone) and relatively low levels of the glucocorticoids downstream of 17-hydroxyprogesterone (i.e., 11-deoxycortisol and cortisol). Laboratory findings associated with this condition include elevated serum and urinary 17-hydroxyprogesterone. ■ In patients with 21-hydroxylase deficiency, shunting of precursor molecules along remaining functional pathways results in a relative increase in dehydroepiandrosterone and androstenedione. In female patients, this phenomenon results in virilization, hirsutism, amenorrhea, and infertility. Manifestations in male patients include precocious puberty and infertility. ■ 11-β-Hydroxylase converts 11-deoxycorticosterone into corticosterone and 11-deoxycortisol into cortisol. Patients with 11-β-hydroxylase deficiency consequently have relatively low levels of corticosterone, aldosterone, and cortisol. Laboratory findings associated with this condition include elevated serum 11-deoxycorticosterone and 11-deoxycortisol. ■ 3-β-Hydroxylase converts pregnenolone into progesterone, 17-hydroxypregnenolone into 17-hydroxyprogesterone, and dehydroepiandrosterone into androstenedione. Patients with 3-β-hydroxylase deficiency consequently have relatively low levels of progesterone (and all mineralocorticoids), 17-hydroxyprogesterone (and all downstream glucocorticoids), and androstenedione. Laboratory findings associated with this condition include elevated serum pregnenolone, 17-hydroxypregnenolone, and dehydroepiandrosterone. ■ Addison’s disease refers to primary adrenal insufficiency. In the United States, this condition most frequently results from autoimmune adrenalitis. ■ AIS, also known as testicular feminization syndrome, causes male pseudohermaphroditism. AIS results from end-organ resistance to androgens, usually caused by qualitative or quantitative abnormalities of the androgen receptor. Androgen biosynthesis and testicular development are normal in patients with this syndrome. The karyotype of these patients is 46,XY.21,43 6a. A. Testosterone. 6b. A. AMH levels are elevated in serum but not in ovarian follicular fluid. 6c. A. Pelvic inflammatory disease (PID). 6d. A. FSH levels in these patients are too high to initiate the maturation process among primordial follicles. Major points of discussion ■ Maturation of follicles, eventually resulting in ovulation, proceeds as follows: During the luteal phase of the menstrual cycle, a cohort of primary follicles begins the maturation process. Each primary follicle consists of a primary oocyte surrounded by a single layer of granulosa cells. Primary follicles mature into secondary/preantral follicles; each secondary follicle has a zona pellucida around its oocyte, surrounded by three to five layers of granulosa cells. Secondary follicles then mature into tertiary/antral/vesicular follicles, each of which contains a fluid-filled antrum and is surrounded by a theca interna and theca externa. Tertiary follicles proceed to become mature/Graafian mature follicles, each of which has an eccentrically located oocyte surrounded by a cumulus oophorus consisting of granulosa cells. ■ By the mid to late luteal phase of each menstrual cycle, fewer than four follicles from that cycle’s cohort persist as mature follicles. The other follicles from each cohort digress into atresia during the maturation process. Of these few mature follicles, only one will become that cycle’s preovulatory follicle. Relative to the other mature follicles, the preovulatory follicle has higher intrafollicular levels of FSH and higher aromatase activity. These properties enable the preovulatory follicle to continue the maturation process, driven by estradiol (synthesized by aromatase) and FSH. ■ Plasma LH levels rise during the late proliferative phase of the endometrial cycle. This change stimulates production of androstenedione by theca cells. Aromatase converts this androstenedione into estradiol, which further drives maturation of the preovulatory follicle. ■ Granulosa cells of early developing follicles produce AMH, also known as Müllerian-inhibiting substance. Although this hormone’s action is incompletely understood, proposed functions include inhibition of initial recruitment of primordial follicles and inhibition of aromatase activity. Accordingly, studies have shown an inverse relationship between serum levels of AMH and estradiol. Studies have also shown an inverse relationship between serum levels of AMH and FSH. ■ In healthy women, serum levels of AMH undergo minor fluctuations during the menstrual cycle, presumably in concert with cyclic growth of small follicles, and loss of follicles via atresia. ■ In patients with PCOS, recruitment and initial growth of follicles proceeds normally, but selection of a dominant preovulatory follicle does not occur. Consequently, the ovaries of these patients contain multiple small Graafian follicles. AMH levels are markedly increased in patients with PCOS. ■ Patients with PCOS are at increased risk for development of endometrial hyperplasia and malignancy as the result of chronic anovulation.14,18,38 7a. A. Granulosa cell tumors secrete steroid hormones; secretion of peptide hormones by these tumors has not been reported. 7b. A. α-Fetoprotein (AFP). 7c. A. Granulosa cell tumors frequently behave aggressively in middle-aged and postmenopausal patients but generally follow a benign course in children and young adults. Major points of discussion ■ Granulosa cell tumors belong to the category of ovarian sex cord−stromal tumors. This category accounts for approximately 8% of ovarian neoplasia. As outlined by the World Health Organization, this category encompasses tumors composed of granulosa cells, theca cells, Sertoli cells, Leydig cells, and fibroblasts of stromal origin. ■ Granulosa cell tumors represent 1% to 2% of ovarian tumors overall and 6% to 10% of ovarian malignancies. ■ The granulosa cell tumor group includes the adult type and the juvenile type. The adult type occurs in middle-aged to postmenopausal women. The granulosa cells have a round to ovoid nucleus with a longitudinal groove. Juvenile granulosa cell tumors are encountered predominantly during the first three decades of life. Almost all nuclei lack grooves. The adult type frequently recurs and behaves invasively, whereas the juvenile type generally follows a benign clinical course. ■ Granulosa cell tumors may produce steroid hormones (e.g., estrogens, progestins) and/or peptide hormones (e.g., inhibin, AMH). ■ Fibromas are the most common sex cord−stromal tumors; these lesions represent approximately 4% of ovarian tumors. They are most common in middle-aged women (patients’ mean age is 48 years). They occur only occasionally in children.30,47 8a. A. Addison’s disease. 8b. A. Elevated plasma renin activity (PRA) and decreased aldosterone. 8c. A. Continued suppression of aldosterone after stimulation with adrenocorticotrotpic hormone. Major points of discussion ■ Hyperaldosteronism should be suspected in patients with refractory hypertension (e.g., receiving more than three anti-hypertensive agents) and in patients with spontaneous hypokalemia (< 3.5 mmol/L), especially when associated with metabolic alkalosis. When patients are treated with diuretics, hyperadrenalism should be suspected when the potassium is less than 3.0 mmol/L and when it fails to normalize 2 to 4 weeks after stopping diuretics. Although potassium can be normal in up to 50% of patients with adrenal hyperplasia, the 24-hour urine potassium levels are almost always higher than 30 mmol/24 hr. ■ Screening tests include simultaneous measurement of plasma aldosterone and renin levels. In the case of primary hyperaldosteronism, the plasma aldosterone will be high-normal or elevated (> 8 to 15 ng/dL), whereas the plasma renin will be low, resulting in a aldosterone/renin ratio higher than 20 to 50 (depending on the laboratory). ■ Plasma aldosterone and renin measurements should be made with careful attention to patient preparation because any condition interfering with plasma volume and the renin-angiotensin-aldosterone-potassium axis can complicate interpretation of the results. Examples include discontinuation of drugs such as spironolactone, ACE inhibitors, diuretics, nonsteroidal anti-inflammatory drugs, β-blockers, and similar agents for at least five half-lives. Other important considerations are correction of hypokalemia with potassium chloride before testing and collection of the blood sample from a seated patient the morning after ambulation for more than 30 minutes. ■ Renin can be measured by its enzymatic activity (PRA) because it cleaves angiotensinogen to form angiotensin 1, which is then measured by an immunoassay. Issues with this approach include avoidance of refrigeration of the sample to prevent cold activation of renin and variability in plasma angiotensinogen levels. Recently, renin mass immunoassays have been developed and will provide a measure of renin levels independent of its activity. ■ Aldosterone immunoassays are also subject to interferences by other sterols, such as in patients with adrenal hyperplasia and elevated levels of 21-deoxyaldosterone, as well as other uncharacterized substances. Measuring aldosterone with a liquid chromatography−mass spectrometry method can largely obviate these issues.46,51,52 9. A. FSH. Major points of discussion ■ The American Society for Reproductive Medicine classification of primary amenorrhea includes three main groups: 1. Primary amenorrhea with normal Tanner stage 2. Primary amenorrhea with low Tanner stage and high FSH level 3. Primary amenorrhea with low Tanner stage and normal or low FSH level ■ In primary amenorrhea with normal Tanner stage, it is important to ascertain whether Müllerian structures (fallopian tubes, uterus, cervix) are present. If the uterus is present, particularly in patients with periodic abdominal pain, outlet abnormalities such as cervical stenosis, vaginal aplasia, vaginal septum, and imperforate hymen are likely. Rarely, primary amenorrhea in a patient with fully developed external female genitalia and normal Müllerian structures with 46,XY karyotype is due to pure gonadal dysgenesis, caused by mutations in genes involved in testis development, including SRY, steroidogenic factor 1 (NR5A1), DAX1 (NR0B1), desert hedgehog (DHH), WT1, WNT4, and SOX9. ■ In primary amenorrhea with normal Tanner stage in the absence of Müllerian structures, a karyotype will distinguish the two most common causes: Müllerian agenesis and complete androgen insensitivity. A normal karyotype and LH and FSH levels within the reference range indicate Müllerian agenesis. Complete androgen insensitivity (testicular feminization), caused by mutations in the androgen receptor, will present with 46,XY karyotype with an external female phenotype with normal breast development, undescended testes, normal or elevated LH and FSH, and testosterone levels in the normal to high male range. ■ Low Tanner staging with elevated LH and FSH indicate delayed puberty caused by hypergonadotropic hypogonadism (gonadal dysgenesis). The most common cause is Turner’s syndrome, defined by a 45,XO karyotype in a female. These patients lose their germ cells during gestation and are at high risk for cardiac, renal, thyroid, and ear abnormalities. Other congenital causes of hypergonadotropic hypogonadism include fragile X, Noonan’s, and Swyer’s syndromes. Causes of acquired primary hypogonadism include premature ovarian failure (idiopathic, trauma, surgery, radiation, chemotherapy, mumps, autoimmune disease); resistant ovary syndrome; and aromatase, 17-hydroxylase, and 17,20-lyase deficiencies. ■ Patients with hypogonadotropic hypogonadism present with low levels of FSH, LH, and estradiol. Elevated prolactin suppresses gonadotropin-releasing hormone and can occur with defects in the hypothalamic-pituitary axis, including pituitary adenomas, brain tumors, infections, hemochromatosis, sarcoidosis, aneurysms, trauma, hypothyroidism, and medications. Other causes include Kallmann’s syndrome (congenital GnRH deficiency), thyroid disease, anorexia nervosa, malnutrition, stress, excessive exercise, and several chronic debilitating diseases. Occasionally, early onset of PCOS, Cushing’s syndrome, ovarian or adrenal tumor, or late-onset congenital adrenal hyperplasia can cause primary amenorrhea with signs of hyperandrogenism such as acne and hirsutism. Hypogonadism with low FSH and LH, a family history of delayed puberty, and exclusion of other diseases can be due to constitutional delay of puberty, when puberty occurs later than 2.5 deviations from the population mean. ■ Physical examination, patient and family history, and laboratory measurements of FSH, LH, and prolactin can identify the most common causes of primary amenorrhea. Depending on the clinical situation, other useful tests include pelvic ultrasonography, head MRI, karyotype, TSH, testosterone, estradiol, DHEA-S, androstenedione, and 17-hydroxyprogesterone.39,58 10. A. Large prolactinoma and hook effect. Major points of discussion ■ Excluding pregnancy, the causes of secondary amenorrhea can be classified into five main groups: 2. Hypergonadotropic hypogonadism (high FSH; about 12%); for example, premature ovarian failure, autoimmune oophoritis, 46,XY karyotype, and fragile X syndrome. 3. Hyperprolactinemia (13%). 4. Anatomic (Asherman’s syndrome, 7%). 5. Hyperandrogenic states (22%); for example, PCOS, ovarian tumors, and late-onset congenital adrenal hyperplasia. ■ The initial laboratory tests for evaluation of secondary amenorrhea should include FSH, LH, TSH, and prolactin levels. ■ Hyperprolactinemia can usually be explained by one of these mechanisms: 2. Decreased dopaminergic inhibition of prolactin secretion: b. Drugs interfering with dopaminergic pathways (e.g., antipsychotics, metoclopramide, methyldopa, reserpine, verapamil, selective serotonin uptake inhibitors). 3. Prolactin-secreting tumor: pituitary lactotroph adenomas. 4. Miscellaneous pathologic causes: hypothyroidism, chest wall injury, chronic renal failure. 5. Macroprolactinemia: prolactin complexed with immunoglobulins (or rarely polymers of prolactin ranging up to 500 kDa) causes decreased renal clearance and elevated concentrations of nonfunctional prolactin. ■ Because a large number of physiologic variables and medications can raise prolactin levels, it is recommended that prolactin testing be repeated to confirm the increased level. It is also recommended to treat the serum with a method that removes macroprolactin (either gel filtration or polyethylene glycol precipitation) in samples with prolactin levels higher than 90 to 100 ng/mL. ■ In women with persistent hyperprolactinemia, the prevalence of a pituitary tumor is approximately 50% to 60%, justifying follow-up with imaging. Microadenomas (< 1 cm) usually have levels below 200 ng/mL, whereas macroadenomas (> 2 cm) typically present with levels above 1000 ng/mL. ■ The hook effect occurs when very high levels of prolactin overwhelm the binding capacity of the reagent antibodies in the immunoassay, causing single antibody-antigen complexes instead of the required “sandwich” (capture antibody-antigen-detection antibody) and a falsely low result. Serial dilution of the antigen before adding the reagents is required to accurately determine the antigen concentration.20,32,39 11. A. Decreased testosterone. Major points of discussion ■ Complete androgen insensitivity (CAIS, testicular feminization) will present with 46,XY karyotype with an external female phenotype, primary amenorrhea, no Müllerian structures (uterus, fallopian tubes, cervix), normal breast development, undescended testes, elevated LH levels, normal or elevated FSH levels, testosterone levels in the normal to high male range, and normal to high estradiol levels. ■ Less severe defects in the androgen receptor cause partial androgen insensitivity (PAIS) with incomplete virilization ranging from female phenotype with clitoromegaly or posterior labial fusion to male phenotype with minor virilization defects such as azoospermia, gynecomastia, and scant facial and pubic hair. In PAIS, abnormalities in sex hormones are less pronounced, and LH, testosterone, and estradiol levels may be normal. ■ Phenotypes similar to PAIS (but without normal breast development) can occur with defects in testosterone synthesis. In these cases, testosterone and estradiol levels are low, LH and FSH levels are elevated with a higher LH/FSH ratio, and precursors proximal to the synthetic defect will be elevated. The most common defect is in HSD17B3, which codes for 17β-hydroxysteroid dehydrogenase. 17β-Hydroxysteroid dehydrogenase converts androstenedione to testosterone in the testes, resulting in accumulation of androstenedione. Most of these patients present with some degree of virilization due to the action of extragonadal androgens. ■ Patients with gonadal dysgenesis and a 46,XY karyotype can also present with a female phenotype. These cases are also characterized by low testosterone and estradiol, high LH/FSH ratio, and low Müllerian inhibitory hormone levels, which result in the formation of normal Müllerian structures. ■ Complete failure of testosterone production can be distinguished from CAIS because postpubertal testosterone will be low in the former and normal to high in the latter. However, partial or incomplete defects are not always easy to distinguish. In prepubertal children with intact testosterone production, a stimulation test with hCG will typically result in an increase in testosterone levels of greater than 200 ng/dL. ■ Laboratory testing for testosterone in children and women should use methods that are sufficiently specific and sensitive at the low concentrations expected in normal individuals; for example, liquid chromatography−mass spectrometry.22,34,39 12. A. CTX reflects significant improvement in osteoporosis, whereas N-terminal PINP does not respond to vitamin D treatment. Major points of discussion ■ Relative to an 8 am sample (fasting or nonfasting), CTX in a sample collected later in the day is lower by up to 50%. Relative to nonfasting, CTX in a sample collected during fasting, especially randomly collected throughout the day, is higher (Figure 4-3, A).
Clinical Chemistry
Endocrine (Thyroid, Pituitary, Adrenal, Bone, Pancreas, Reproductive) and Catecholamines
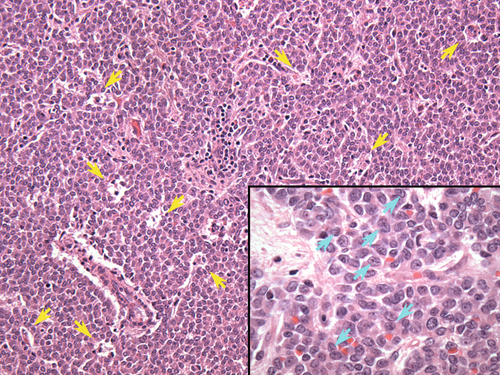
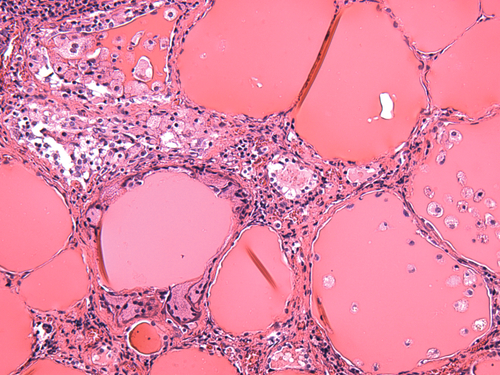
Rationale: This is the definition of suppression in the high-dose DST and would be seen in patients with Cushing’s disease (secondary hyperadrenalism).
B. No suppression of serum cortisol.
Rationale: The high-dose DST is used to differentiate the cause of Cushing’s syndrome as ACTH dependent or independent. Given the patient’s CT finding, the patient most likely has an adrenal adenoma or carcinoma and the Cushing’s syndrome is ACTH independent. The patient’s serum cortisol will therefore not be suppressed with high-dose dexamethasone.
C. An elevation in serum cortisol.
Rationale: An elevation in serum cortisol is possible but is not generally seen in the DST.
D. Suppression of serum metanephrines.
Rationale: Serum metanephrines would not be expected to change with the DST because they are not under the control of ACTH.
B. Normal TSH, elevated FT4, elevated FT3.
C. Elevated TSH, decreased FT4, decreased FT3.
D. Elevated TSH, normal FT4, normal FT3.
E. Elevated TSH, elevated FT4, elevated FT3.
Rationale: Reduced end-organ responsiveness to thyroid hormone would result in a compensatory increase in TSH, FT4, and FT3.
Rationale: This patient’s goiter likely results from increased stimulation of the thyroid by TSH, which in turn results from reduced end-organ responsiveness to thyroid hormone. This mutation would not lead to increased TSH and would not affect end-organ responsiveness to thyroid hormone.
B. Inactivating mutation of the thyroid stimulating hormone receptor (TSHR) gene.
Rationale: This mutation would decrease the effectiveness of TSH (i.e., TSH would bind to a nonfunctional TSHR) and would not affect end-organ responsiveness to thyroid hormone.
C. Inactivating mutation of the TSH β-chain gene.
Rationale: This mutation would lead to a decrease in the functionality of TSH and would not affect end-organ responsiveness to thyroid hormone.
D. Inactivating mutation of the thyroxine-binding globulin (TBG) gene.
Rationale: This mutation would not lead to increased TSH and would not affect end-organ responsiveness to thyroid hormone.
E. Inactivating mutation of the thyroid hormone receptor (TR) β gene.
Rationale: In the context of this mutation, end-organ responsiveness to thyroid hormone would be reduced. Goiter would form as a result of a compensatory increase in TSH.
Rationale: T3 is more metabolically active than T4.
B. T4 is more metabolically active than rT3, which is more metabolically active than T3.
C. T3 and rT3 are approximately equally metabolically active. These hormones are more metabolically active than T4.
D. T3 is more metabolically active than T4, which is more metabolically active than rT3.
E. T3 is more metabolically active than rT3, which is more metabolically active than T4.
Rationale: T3 is the most metabolically active of these hormones, followed by T4 and then rT3.
Rationale: Liver is among the most thyroid-responsive tissues.
B. Kidney.
Rationale: Kidney is among the most thyroid-responsive tissues.
C. Spleen.
Rationale: Spleen is considered relatively thyroid unresponsive.
D. Skeletal muscle.
Rationale: Skeletal muscle is among the most thyroid-responsive tissues.
E. Heart.
Rationale: Heart is among the most thyroid-responsive tissues.
Rationale: This patient exhibits features of hypogonadism, likely secondary to chemotherapy during his childhood. Men with hypogonadism that began before puberty typically demonstrate a female fat distribution pattern, which includes increased fat deposition in the hips, buttocks, and thighs.
B. Increased fat deposition in the face, abdomen, and waist.
Rationale: Men with hypogonadism that began before puberty typically demonstrate a female fat distribution pattern, which includes decreased fat deposition in the face, abdomen, and waist.
C. Decreased facial and body hair.
Rationale: Decreased facial and body hair are typical findings in men with hypogonadism.
D. Decreased hair on the temporal aspect of the scalp.
Rationale: Men with hypogonadism typically do not undergo temporal hair recession.
E. A diamond-shaped escutcheon (pubic hair pattern), rather than a triangular escutcheon.
Rationale: Men with hypogonadism that began before puberty typically demonstrate a female (inverted triangular) escutcheon, rather than a male (diamond-shaped) escutcheon.
Rationale: Klinefelter’s syndrome is the most common genetic cause of primary hypogonadism in men.
B. XYY syndrome.
Rationale: Testosterone levels are normal in 47, XYY males.
C. Kallmann’s syndrome.
Rationale: Kallmann’s syndrome (anosmic hypogonadotropic hypogonadism) is a cause of central hypogonadism.
D. Loss-of-function mutations in the gene encoding GPR54.
Rationale: These mutations are associated with idiopathic hypogonadotropic hypogonadism.
E. CHARGE syndrome.
Rationale: Mutations of the gene CHD7, which is associated with the CHARGE syndrome (coloboma, heart anomalies, choanal atresia, retardation of growth and development, and genital and ear anomalies), have been identified in patients with hypogonadism.
B. Estrogens ultimately terminate growth by causing closure of the epiphyses of the long bones.
Rationale: Estrogens are derived from androgens. Patients with prepubertal hypogonadism undergo delayed epiphyseal closure and consequently have tall stature.
C. Androgens have a net anabolic effect in terms of protein metabolism (i.e., androgens increase protein synthesis and decrease protein degradation).
D. Androgens increase the deposition of calcium salts and the rate of bone growth.
Rationale for A, C, and D: These statements are true but do not explain the tall stature of a patient with decreased androgen levels.
E. Boys who undergo precocious puberty (i.e., onset of puberty before age 9 years) typically have tall stature as adults.
Rationale: Boys who undergo precocious puberty typically have short stature as adults. Estrogens, which are derived from androgens, ultimately terminate growth by causing closure of the epiphyses of the long bones. Boys with precocious puberty, therefore, undergo premature epiphyseal closure.
Rationale: Serum total testosterone levels and serum free testosterone levels decrease with age.
B. The quantitative relationship between serum total testosterone and serum free testosterone depends primarily on the serum concentration of albumin.
Rationale: The quantitative relationship between serum total testosterone and serum free testosterone levels depends primarily on the serum concentration of sex hormone−binding globulin.
C. Serum testosterone levels are inversely proportional to body mass index (BMI).
Rationale: Cross-sectional epidemiologic studies have shown an inverse relationship between BMI and circulating testosterone levels.
D. Serum testosterone levels have no quantitative relationship with central obesity (i.e., waist/hip ratio).
Rationale: Cross-sectional epidemiologic studies have shown an inverse relationship between central obesity and circulating testosterone levels.
E. Serum total testosterone levels are higher in young white men compared with young black men.
Rationale: Results of studies comparing serum testosterone levels of white and black men have been mixed. Some studies demonstrate that serum testosterone levels are higher in black men compared with white men, especially among younger adult men, whereas other studies have not shown any difference between the two groups even at younger ages.
Rationale: Pheochromocytomas associated with Cushing’s syndrome are well-documented entities.
B. The increased urine free cortisol indicates a functional adrenal cortical neoplasm and effectively rules out the possibility of a malignant neoplasm.
Rationale: Functional tumors are not necessarily benign. Functional adrenal cortical carcinomas and catecholamine-producing malignant pheochromocytomas are well-documented entities.
C. Cushing’s syndrome caused by ectopic production of ACTH occurs more frequently than ACTH-independent Cushing’s syndrome.
Rationale: ACTH-independent Cushing’s syndrome caused by adrenal tumors is approximately twice as common as ACTH-dependent Cushing’s syndrome caused by ectopic production of ACTH.
D. The patient’s psychosis is probably related to her adrenal mass.
Rationale: Clinical features of Cushing’s syndrome include various psychiatric symptoms.
E. The patient’s hypokalemia is probably due to increased intracellular potassium uptake caused by high serum cortisol levels.
Rationale: Cortisol possesses some mineralocorticoid activity. In the context of Cushing’s syndrome, hypokalemia results from increased renal secretion of potassium mediated by cortisol.
Rationale: Pregnenolone is a biosynthetic precursor of cortisol.
B. Progesterone.
Rationale: Progesterone is a biosynthetic precursor of cortisol.
C. 11-Deoxycortisol.
Rationale: 11-Deoxycortisol is a glucocorticoid precursor.
D. Corticosterone.
Rationale: Corticosterone is a mineralocorticoid precursor, rather than a glucocorticoid precursor.
E. Cortisone.
Rationale: Cortisone is a glucocorticoid (like cortisol) and exists in equilibrium with cortisol.
Rationale: Cortisol increases hepatic gluconeogenesis by inhibiting protein synthesis, thereby increasing the amounts of amino acid substrates for gluconeogenesis.
B. Cortisol decreases hepatic gluconeogenesis by accelerating protein degradation, thereby increasing the amounts of amino acid substrates for gluconeogenesis.
Rationale: Cortisol increases hepatic gluconeogenesis by accelerating protein degradation, thereby increasing the amounts of amino acid substrates for gluconeogenesis.
C. Cortisol directly activates key hepatic gluconeogenic enzymes and increases the activity of hepatic glycogen synthetase.
Rationale: Cortisol increases glycogen stores in the liver, or gluconeogenesis, and decreases uptake of glucose by peripheral tissues, leading to hyperglycemia.
D. Cortisol increases the uptake and phosphorylation of glucose by peripheral tissues.
Rationale: Cortisol decreases the uptake and phosphorylation of glucose by peripheral tissues.
Rationale: Primary hyperaldosteronism (Conn’s syndrome) is characterized by hypokalemia and hypertension (among other features) and is almost always due to an adenoma of the adrenal cortex.
B. Virilizing tumors of the adrenal gland occur rarely, cause clinical changes in women but not in men, and are associated with low levels of dehydroepiandrosterone sulfate (DHEA-S) in serum and 17-ketosteroids in urine.
Rationale: Virilizing tumors of the adrenal gland occur rarely, cause clinical changes in women but not in men, and are associated with high levels of DHEA-S in serum and 17-ketosteroids in urine.
C. Pheochromocytomas are associated with multiple endocrine neoplasia syndrome, types IIa (MEN IIa) and IIb (MEN IIb), von Hippel–Lindau syndrome, familial paraganglioma syndrome, and neurofibromatosis I.
Rationale: All answer choices are correct.
D. Neuroblastoma is a well-differentiated tumor of childhood that rarely produces catecholamines.
Rationale: Neuroblastoma is a poorly differentiated tumor of childhood, and catecholamine production is seen in more than 90% of cases.
Rationale: 17-Hydroxyprogesterone levels would not be increased in a patient with Addison’s disease.
B. 21-Hydroxylase deficiency.
Rationale: 21-Hydroxylase converts progesterone into 11-deoxycorticosterone (a precursor of aldosterone) and 17-hydroxyprogesterone into 11-deoxycortisol (a precursor of cortisol). Patients with 21-hydroxylase deficiency have markedly increased levels of serum 17-hydroxyprogesterone and reduced levels of serum 11-deoxycortisol.
C. 11-β-Hydroxylase deficiency.
Rationale: Patients with 11-β-hydroxylase deficiency have increased levels of serum 11-deoxycortisol.
D. 3-β-Hydroxylase deficiency.
Rationale: 17-Hydroxyprogesterone levels would not be increased in a patient with 3-β-hydroxylase deficiency.
E. Androgen insensitivity syndrome (AIS).
Rationale: Normal male genitalia are inconsistent with AIS.
Rationale: 17-Hydroxypregnenolone is a precursor of 17-hydroxyprogesterone.
B. Aldosterone.
C. 18-Hydroxycorticosterone.
D. Corticosterone.
Rationale: Patients with 21-hydroxylase deficiency have relatively low levels of mineralocorticoids (which are downstream of progesterone).
E. Cortisol.
Rationale: Patients with 21-hydroxylase deficiency have relatively low levels of the glucocorticoids downstream of 17-hydroxyprogesterone (i.e., 11-deoxycortisol and cortisol).
Rationale: Serum levels of pregnenolone would likely be increased in a patient with 21-hydroxylase deficiency.
B. Increased serum levels of 11-deoxycorticosterone.
Rationale: Serum levels of 11-deoxycorticosterone would likely be decreased in a patient with 21-hydroxylase deficiency.
C. Decreased serum levels of dehydroepiandrosterone.
Rationale: Serum levels of dehydroepiandrosterone would likely be increased in a patient with 21-hydroxylase deficiency.
D. Decreased serum levels of progesterone.
Rationale: Serum levels of progesterone would likely be increased in a patient with 21-hydroxylase deficiency.
E. An abnormal physical examination.
Rationale: In female patients with 21-hydroxylase deficiency, high levels of dehydroepiandrosterone and androstenedione may cause virilization and hirsutism.
Rationale: Testosterone levels are generally elevated in patients with PCOS.
B. Androstenedione.
Rationale: Androstenedione levels are generally elevated in patients with PCOS.
C. Luteinizing hormone (LH).
Rationale: LH levels are generally elevated in patients with PCOS.
D. Follicle-stimulating hormone (FSH).
Rationale: FSH levels are usually within normal limits in PCOS but may be low in some cases.
E. Insulin.
Rationale: Insulin levels are generally elevated in patients with PCOS because of the strong association between PCOS and insulin resistance.
B. AMH levels are elevated in ovarian follicular fluid but not in serum.
C. AMH levels are elevated in serum and in ovarian follicular fluid.
D. AMH levels are not elevated in serum or ovarian follicular fluid.
Rationale: In patients with PCOS, AMH levels are elevated in serum and in ovarian follicular fluid.
E. In patients with PCOS, the abnormality of AMH is qualitative rather than quantitative.
Rationale: In patients with PCOS, AMH is qualitatively normal.
B. Salpingitis isthmica nodosa (SIN).
C. Endometriosis.
Rationale: Endometriosis is not associated with increased levels of AMH in serum or ovarian follicular fluid.
D. Pelvic adhesions from prior surgery.
Rationale for A, B, and D: Infertility due to “tubal factors” (e.g., PID, SIN, or pelvic adhesions) is not associated with increased levels of AMH in serum or ovarian follicular fluid.
E. Granulosa cell tumors.
Rationale: AMH levels may be very high in patients with granulosa cell tumors.
Rationale: FSH levels in these patients are too low to initiate the maturation process among primordial follicles.
B. Low serum LH levels prevent primordial follicles from initiating the process of maturation.
Rationale: High serum LH levels prevent primordial follicles from initiating the process of maturation.
C. AMH activity prevents primordial follicles from initiating the process of maturation.
Rationale: AMH levels may be markedly increased in PCOS.
D. AMH stimulates aromatase activity within granulosa cells, thereby reducing production of estradiol.
Rationale: AMH inhibits aromatase activity within granulosa cells, thereby reducing production of estradiol.
B. Granulosa cell tumors secrete peptide hormones; secretion of steroid hormones by these tumors has not been reported.
C. Granulosa cell tumors secrete steroid hormones (including estrogen, testosterone, progestins, and inhibin), as well as peptide hormones (including AMH).
Rationale: Inhibin is a peptide hormone, not a steroid hormone.
D. Granulosa cell tumors secrete steroid hormones (including estrogen, testosterone, and progestins), as well as peptide hormones (including AMH and inhibin).
E. Granulosa cell tumors are generally poorly differentiated neoplasms, and production of functional hormones by these lesions is exceedingly rare.
Rationale: Secretion of steroid hormones (e.g., estrogens, progestins) and/or peptide hormones (e.g., inhibin, AMH) by granulosa cell tumors is a well-recognized phenomenon.
Rationale: Increased AFP has been reported in the context of juvenile granulosa cell tumor.
B. β-Human chorionic gonadotropin (β-hCG).
Rationale: β-hCG levels are generally normal in patients with granulosa cell tumors.
C. Carcinoembryonic antigen (CEA).
Rationale: CEA levels are generally normal in patients with granulosa cell tumors.
D. Lactate dehydrogenase (LDH).
Rationale: LDH levels are generally normal in patients with granulosa cell tumors.
E. CA-125.
Rationale: CA-125 levels are generally normal in patients with granulosa cell tumors.
B. Granulosa cell tumors frequently behave aggressively in children and young adults but generally follow a benign course in middle-aged and postmenopausal patients.
C. Granulosa cell tumors frequently behave aggressively in younger and older patients alike.
D. Granulosa cell tumors generally follow a benign course in younger and older patients alike.
E. Granulosa cell tumors generally follow a benign course in middle-aged and postmenopausal patients; these tumors have not been described in children and young adults.
Rationale: The granulosa cell tumor group includes the adult type and the juvenile type. The adult type occurs in middle-aged to postmenopausal women and behaves aggressively. Juvenile granulosa cell tumors are encountered predominantly during the first three decades of life and generally follow a benign clinical course.
Rationale: Hypoadrenalism leads to hypotension, hyperkalemia, and metabolic acidosis.
B. Graves’ disease.
Rationale: Hyperthyroidism can be associated with hypertension but usually not hypokalemia because of renal loss.
C. Conn’s syndrome.
Rationale: Hyperaldosteronism causes renal loss of potassium and protons. resulting in hypokalemia and hypochloremic metabolic alkalosis.
D. Mild congenital adrenal hyperplasia.
Rationale: Deficiency in adrenocorticoid synthesis pathways can lead to hypoaldosteronism, typically compensated in mild cases by increased ACTH and consequent adrenal hyperplasia.
E. Panhypopituitarism.
Rationale: Pituitary or hypothalamic lesions can lead to secondary or tertiary adrenal insufficiency, not hyperadrenalism.
Rationale: Primary hypoaldosteronism can result in hypotension and compensatory increased renin.
B. Suppressed PRA and elevated aldosterone.
Rationale: Primary hyperaldosteronism leads to salt retention, expanded blood volume, hypertension, and feedback suppression of renin production by the juxtaglomerular apparatus.
C. Elevated 17-hydroxyprogesterone and low aldosterone.
Rationale: These are findings consistent with congenital adrenal hyperplasia caused by defective 21-hydroxylase (CYP21A2) or 11-β-hydroxylase (CYP11B1/B2).
D. Elevated FT4 and suppressed TSH.
Rationale: These are findings in Graves disease.
E. Low TSH and low FT4.
Rationale: These are consistent with secondary hypothyroidism.
Rationale: Inability to produce aldosterone after multiday ACTH stimulation is consistent with primary hypoaldosteronism (e.g., Addison’s disease).
B. Failure to increase TSH after stimulation with thyroid-releasing hormone.
Rationale: This is a response consistent with pituitary insufficiency.
C. Maintained aldosterone elevation after infusion of 2000 mL of isotonic saline.
Rationale: A confirmatory test for Conn’s syndrome is failure to suppress aldosterone production (plasma aldosterone > 10 ng/dL) after expansion of intravascular volume with infusion of 300 to 2000 mL of isotonic saline over a period of 4 hours, indicating autonomous aldosterone production.
D. Elevated 17-hydroxyprogesterone after ACTH stimulation.
Rationale: This is consistent with congenital adrenal hyperplasia.
E. Failure to increase cortisol after insulin-induced hypoglycemia.
Rationale: This is consistent with secondary or tertiary hypoadrenalism.
Rationale: FSH and LH levels establish whether primary amenorrhea is caused by failure of the gonads (levels are high) or hypothalamic-pituitary axis (levels are low).
B. DHEA-S.
Rationale: DHEA-S may be elevated in PCOS and other conditions associated with hyperandrogenism, which accounts for less than 5% of cases of primary amenorrhea.
C. Androstenedione.
Rationale: Androstenedione may be elevated in PCOS and other conditions associated with hyperandrogenism and hirsutism, which accounts for less than 5% of cases of primary amenorrhea.
D. Progesterone.
Rationale: Progesterone levels are not usually helpful in the diagnosis of primary amenorrhea.
E. β-hCG.
Rationale: Pregnancy is a major cause of secondary amenorrhea but not primary amenorrhea.
Rationale: Given the discrepancy between her prolactin levels and a biopsy-confirmed prolactin-secreting macroadenoma, the serum was repeatedly diluted and revealed a prolactin level of 8000 ng/mL.
B. Non–prolactin-secreting tumor and macroprolactin.
Rationale: Macroprolactin usually presents with elevations greater than 90 ng/mL. Tumors and lesions affecting the hypothalamus can cause mild hyperprolactinemia by interfering with the dopaminergic inhibition of prolactin secretion.
C. Stress-associated hyperprolactinemia.
Rationale: Stress can lead to mild hyperprolactinemia, rarely exceeding 40 ng/mL, but is insufficient to explain all the patient’s findings.
D. Graves’ disease.
Rationale: Hypothyroidism, but not hyperthyroidism, is frequently associated with hyperprolactinemia and amenorrhea.
E. Unreported opiate use.
Rationale: Opiates can interfere with dopaminergic pathways and lead to mild hyperprolactinemia, but this is insufficient to explain all the patient’s findings.
Rationale: This patient has AIS, which is characterized by testosterone and other androgens in the normal to elevated male range with a female phenotype.
B. Decreased LH and FSH.
Rationale: These tend to be elevated in AIS because feedback inhibition by androgens is absent.
C. Low estradiol.
Rationale: There is no ovarian hormone production, but elevated testosterone is converted to estradiol by aromatase in extragonadal tissues. These patients can present with normal testosterone and elevated estradiol.
D. Elevated 17-hydroxyprogesterone.
Rationale: This is seen in a form of congenital adrenal hyperplasia with elevated adrenal androgens.
E. Mutated androgen receptor.
Rationale: This is the cause of AIS.
Rationale: Vitamin D improves bone health and alters levels of bone turnover markers. However, the patient was taking vitamin D supplements for more than 2 years during both occasions, so the described change is unlikely to be related to vitamin D treatment.
B. CTX reflects significant improvement in osteoporosis, whereas PINP is falsely elevated because of the influence of food intake and circadian variability on PINP levels.
Rationale: The influence of food intake on N-terminal PINP levels is minimal. Circadian variability in PINP levels is low.
C. The difference between the two CTX levels is within the expected analytical variability.
Rationale: The minimum significant change for CTX measured by CLIA is 27%.
D. Influence of food intake on CTX levels and circadian variability in CTX levels.
Rationale: Fasting substantially increases CTX levels. During fasting, there is also substantial circadian variability in CTX levels with high levels in the morning, followed by decreasing levels during the day.
E. Intralaboratory variability in CTX and PINP results.
Rationale: Intralaboratory variability using automated CLIA is low. Interlaboratory variability can be high.
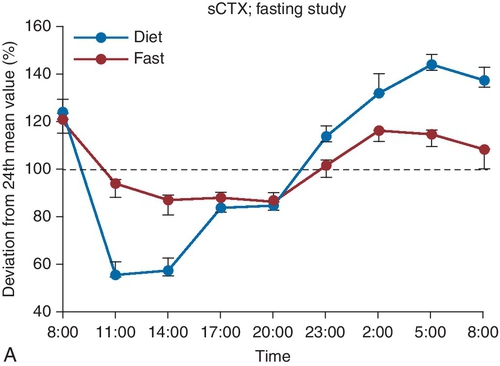
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Clinical Chemistry: Endocrine (Thyroid, Pituitary, Adrenal, Bone, Pancreas, Reproductive) and Catecholamines
Figure 4-1 Micrograph showing the characteristic features of an adult-type granulosa cell tumor. The lesion consists of neoplastic granulosa cells that proliferate in a microcystic pattern, forming Call-Exner bodies (yellow arrows). The inset shows the tumor’s cytomorphology. The tumoral granulosa cells have scant cytoplasm and ovoid nuclei with characteristic longitudinal grooves (blue arrows).
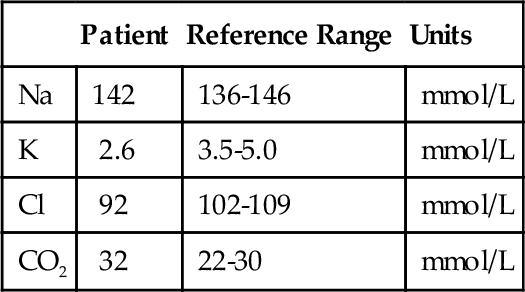
Figure 4-2 Representative section from the thyroid of a 67-year-old man after total thyroidectomy. This section of thyroid shows follicles (some of which are disrupted) associated with foamy macrophages.
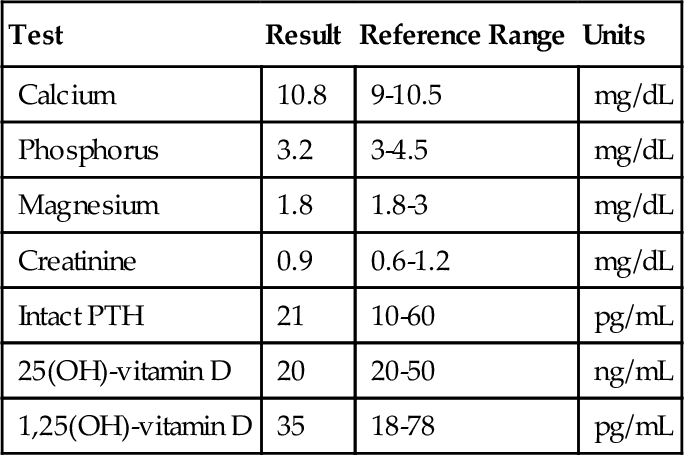
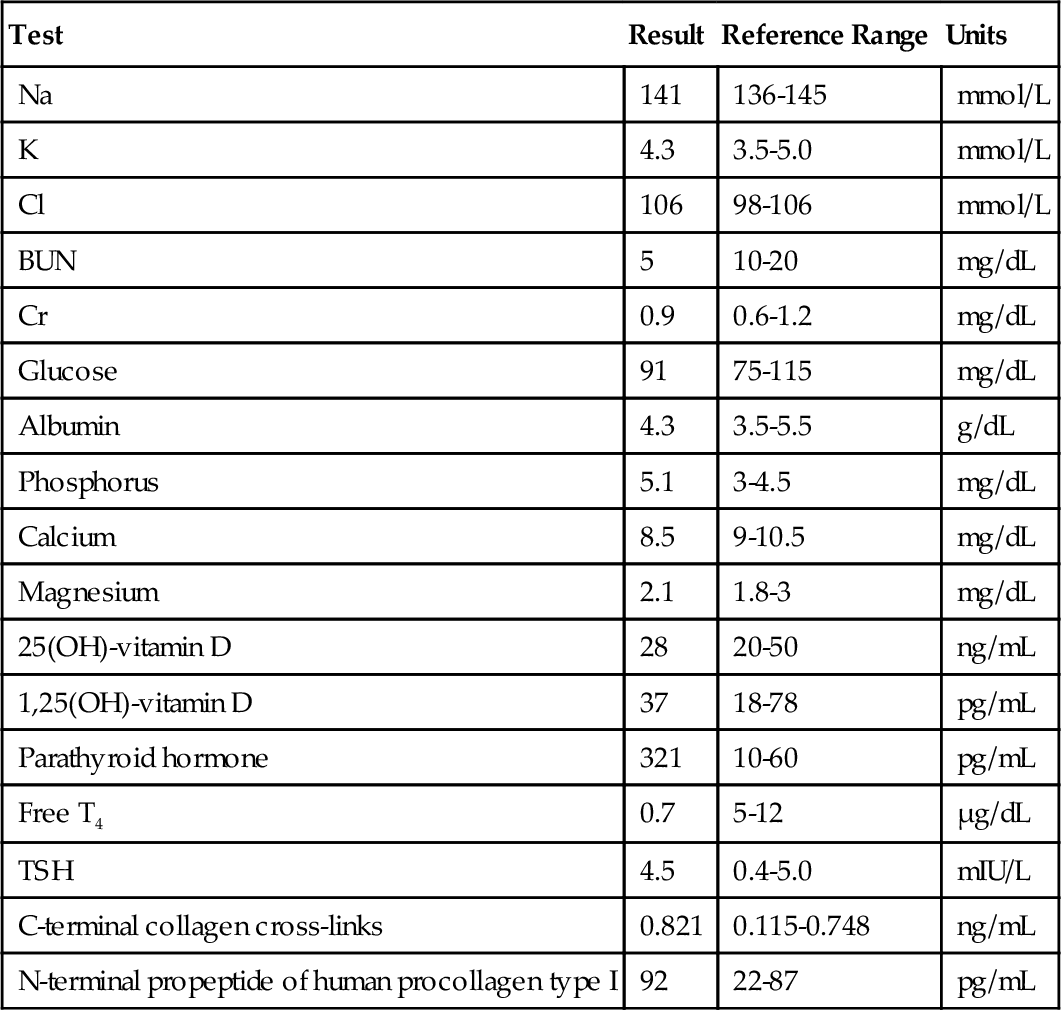
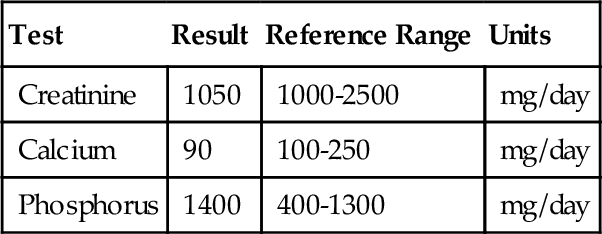
Figure 4-3 A, Serum carboxy-terminal collagen cross-links (sCTX) in premenopausal women on a normal diet and during fasting. B, The effect of feeding on bone turnover markers in individual subjects as a percentage difference (fed minus fasting). Each circle represents one subject. The horizontal line indicates the mean percentage difference for each marker. (A, From Qvist P, Christgau S, Pedersen BJ, et al. Circadian variation in the serum concentration of C-terminal telopeptide of type I collagen (serum CTx): effects of gender, age, menopausal status, posture, daylight, serum cortisol, and fasting. Bone 2002;31[1]:57−61. B, From Clowes JA, Hannon RA, Yap TS, et al. Effect of feeding on bone turnover markers and its impact on biological variability of measurements. Bone 2002;30[6]:886−890.)
