Chronic Disorders of Neurologic Function
Joni D. Marsh
Key Questions
• How are the various types of seizures recognized, classified, and treated?
• How is Alzheimer dementia diagnosed and managed?
• What are the similarities between Alzheimer dementia and vascular dementia?

http://evolve.elsevier.com/Copstead/
Patients experiencing neurologic dysfunction from chronic disease states present a challenge to health care professionals, who must strive to maximize the patient’s function and quality of life. This chapter focuses on common chronic disabilities of neurologic function including those primarily affecting the brain such as seizures, dementia, Parkinson disease, cerebral palsy, and hydrocephalus. Disorders of the spinal cord or peripheral nervous system include multiple sclerosis (MS), spina bifida, and spinal cord injury. Guillain-Barré syndrome and Bell palsy are examples of disorders affecting the peripheral nervous system.
Brain and Cerebellar Disorders
Seizure Disorder
Seizures are a transient neurologic event of paroxysmal abnormal or excessive cortical electrical discharges that are manifested by disturbances of skeletal motor function, sensation, autonomic visceral function, behavior, or consciousness. Symptoms are not constant, and the length of time between seizure episodes is extremely variable. A seizure may only occur once in a person’s lifetime. Epilepsy or seizure disorder refers to recurrent seizures. Seizures are a component of many diseases. Epilepsy affects 2 million Americans. It is predicted that 44 per 100,000 new cases will be diagnosed each year.1
Etiology
Seizures have many causes, and under the right circumstances anyone can experience a seizure. A seizure disorder can be acquired as a consequence of cerebral injury or other pathologic process, including structural lesions such as tumors, blood clots, or infection. Other causes include metabolic and nutritional disorders such as electrolyte and water imbalance, hypoxia, acidosis, pyridoxine deficiency, acute withdrawal from alcohol, therapeutic medication overdose or medication adverse effect, and exposure to toxins such as heavy metals or street drugs. If seizures develop as a result of a structural change such as head injury or stroke, the onset is not predictable. In some cases, seizures may not develop for months or years after the structural change has occurred. In some cases, no explanation for the seizure disorder can be found. These individuals are classified as having idiopathic seizures.
A seizure event is often triggered by specific stimuli, usually unique for each individual. Physical inducements include specific sensory stimuli such as flashing lights, loud noises, and rhythmic music. Fever, physical exhaustion, sleep deprivation, fatigue, inadequate nutrition, hormonal changes of the menstrual cycle, hyperventilation, injury, and drugs can also prompt seizure activity. Psychosocial factors include family and environmental stress, shock, and emotional stress.
Pathogenesis
Seizures are due to an alteration in membrane potential that makes certain neurons abnormally hyperactive and hypersensitive to changes in their environment. These physiologically abnormal neurons form an epileptogenic focus (i.e., an area of the brain from which the seizure emanates). The epileptogenic focus functions autonomously, emitting excessively large numbers of paroxysmal electrical discharges. Results from animal studies suggest that neuroinflammation may be a cause or consequence of these electrical abnormalities.2 Nerve cells in this area can recruit neurons in adjacent areas as well as synaptically related neurons in distant areas of the brain, greatly increasing the number of neurons involved in the seizure activity. Recruitment can also incorporate neurons in the opposite hemisphere. Clinical symptoms become evident when a sufficient number of neurons have been excited. Seizures are classified according to clinical symptoms and the electroencephalographic (EEG) features. Clinical manifestations depend on the area of the brain involved, the area of origin, and the areas to which the seizure spreads.
Clinical manifestations
Seizures may be classified as partial, in which only part of the brain surface is affected (also known as focal seizures), or generalized, in which the whole brain surface is affected during the seizure (Box 45-1).
Generalized seizures
Episodes in which the entire brain is involved from the onset of the seizure are referred to as generalized seizures. Involvement of the thalamus and reticular activating system results in loss of consciousness. Metabolic or toxin-induced seizures tend to be generalized. This category includes the following: absence (petite mal), atypical absence, myoclonic, atonic (drop attack), or tonic-clonic (grand mal) seizures.
Absence or petite mal seizures usually occur only in children and are sometimes identified in children manifesting poor academic performance. They are very brief (2 to 10 seconds), and episodes are characterized by staring spells that last only seconds. Onset and termination of attacks are abrupt. During the spell, the individual is unaware of the surrounding environment and is usually motionless; however, it is not unusual for the person to continue walking or performing a routine motor task. If the seizure activity occurs during conversation, the individual may pause or miss a few words. Absence seizures almost always occur during childhood and resolve by age 20 years, although another seizure type may occur later in life.3 Atypical absence seizures have accompanying myoclonic jerks and automatisms (such as lip smacking or repetitive semi–purposeful movements) with the staring spell. The electroencephalographic patterns are unique to each syndrome. Myoclonic seizures are extremely brief and are characterized by a single jerk or multiple jerks of one or more muscle groups. Atonic seizures or drop attacks are characterized by a sudden and complete loss of muscle tone. Falls and injuries are common with this type of seizure activity. Myoclonic episodes may also be associated with atonic seizures. Tonic-clonic seizures involve stiffening and repetitive jerking of muscle groups.
Tonic-clonic or grand mal seizures are characterized by a sudden loss of consciousness followed by muscle rigidity (tonic phase). The individual falls, and initial motor signs include opening of the mouth and eyes, extension of the legs, and adduction of the arms. There may be tongue biting or a high-pitched cry while the whole musculature is in spasm and air is forced out of the lungs through closed vocal cords. Respiration is arrested, and cyanosis may occur. Bowel and bladder incontinence frequently occurs. The tonic phase may last 10 to 15 seconds and is followed by clonic activity, in which there is often violent but rhythmic muscular contractions. During this phase the eyes roll, the face grimaces, and the pulse rate accelerates. Salivation increases and the patient may become diaphoretic. The clonic phase usually lasts 1 to 2 minutes with a gradual decline in the amplitude of the clonic jerks. The individual remains apneic until the end of the clonic phase that is marked by a deep inspiration.
During the terminal or postictal phase, the individual may regain consciousness or drift into a deep coma-like state. Disorientation and confusion are common. If allowed, the individual may sleep for several hours. Other findings include headache, drowsiness, nausea, muscle soreness, no memory of the seizure event, and retrograde amnesia. During the seizure, the person is at risk for injury from the initial fall as well as from the muscle contractions of the clonic phase.
A potentially life-threatening situation known as status epilepticus occurs in some seizure disorders. Status epilepticus is a continuing series of seizures without a period of recovery between seizure episodes. It can occur with all types of seizures but is of greatest concern in tonic-clonic seizures. Irreversible brain damage and possible death from hypoxia, cardiac dysrhythmias, or lactic acidosis can occur if the airway is not maintained and seizure activity is not halted. Whether nonconvulsive status epilepticus causes neuronal damage is still a matter of debate. Studies of elderly patients with nonconvulsive status epilepticus show very high mortality rates and ongoing research is suggesting that EEG be part of the evaluation for patients with altered levels of consciousness that cannot be otherwise explained.3,4
Partial seizures
Partial seizures are those in which activity is restricted to one brain hemisphere. They are further divided into three categories: simple partial, complex partial, and partial seizures that are secondarily generalized.
In simple partial seizures, the individual does not have a change in level of consciousness. The symptoms may be motor, sensory, or autonomic, or any combination of the three. Motor symptoms may be limited to one part of the body. Sensory seizures may result in tingling or numbness that spreads or “marches” to different parts of the limb or body (depending on the location of the seizure activity in the brain) or may involve the special senses, producing auditory (buzzing sounds), olfactory, or visual manifestations (flashing lights). Autonomic symptoms may include pupillary (pupil dilation), skin (diaphoresis, flushing), or respiratory changes.
Complex partial seizures have many different combinations of cognitive, affective, and psychomotor symptoms. Either loss or alteration of consciousness may occur when the seizure begins. After the attack, the individual may feel drowsy or confused. At the onset of impairment of consciousness, the individual often displays automatisms. Aggressive behavior may be displayed as well, especially if bystanders attempt to restrain the individual. Complex partial seizures often last several minutes and may be followed by a postictal state.
Partial seizures that are secondarily generalized are the third subtype of partial seizures. This category comprises seizures that begin as simple partial seizures and then progress to involve both brain hemispheres. Once generalized, these seizures are clinically similar to primary generalized seizures.
Aura/prodrome
Some people may have a subjective sense of an impending seizure. This prodromal period may be characterized by any one of several phenomena such as a type of myoclonic jerking, headache, lethargy, mood alterations, palpitations, or epigastric sensations, which may precede the actual seizure by several hours. In about half of cases there is some type of movement or odd sensory experience (visual, auditory, olfactory, or gustatory) that occurs seconds before consciousness is lost and that is remembered by the individual after recovery from the seizure. This experience is known as an aura. Although the individual may interpret the aura as an indication that a seizure is about to occur, in fact it is the beginning of the seizure episode. Auras can be significant, because they may be a clue to the location of the epileptogenic focus.
Diagnosis and treatment
The diagnosis and management of seizure disorders are based on the patient’s history, physical, and neurologic examination results as well as the results of electroencephalographic studies. Electroencephalograms (EEGs) may be normal between seizures, so activation techniques (sleep deprivation, hyperventilation) may be used to elicit the pathologic mechanism. Laboratory studies are frequently used to investigate possible metabolic abnormalities as well as therapeutic serum levels in those already using anticonvulsant drugs. Lumbar puncture may be utilized when there is a suspicion of a central nervous system (CNS) infection.3 Initial studies ruling out structural causes may include computed tomography (CT) or magnetic resonance imaging (MRI).
Treatment of an individual experiencing a seizure is concentrated on maintaining an airway and protecting the individual from injury. Recording the course of the seizure episode is useful for identifying the location of the epileptogenic focus and for noting any change in the patient’s seizure pattern. These data are useful in treatment planning. The information recorded should include the time of onset and duration of the seizure, precipitating factors, presence of a prodrome or aura, sequence of seizure activity, autonomic signs, level of consciousness, and postictal state.
Long-term treatment depends on the cause of the seizure disorder. In seizures resulting from a metabolic abnormality, infection, or tumor, the precipitating source is removed. If the seizures are due to irreversible or unidentifiable factors, anticonvulsant medications specific to the type of seizure are the best management. The decision to treat after one seizure is controversial when an identifiable cause has not been found.5 The objective of therapy is to achieve seizure control with a minimum of side effects. Medication is continued until there have been no seizures for at least 2 years and is then gradually withdrawn.3 If seizures continue despite treatment at a maximal dose of a single medication, a second agent is added and the dosage is increased depending on patient tolerance. The first drug is then gradually discontinued. Anticonvulsant medication is a form of control, not a cure.
Treatment also includes patient education in the avoidance of activating factors (e.g., stress, loud noise, alcohol). Patients should be advised to avoid situations that could be dangerous or life threatening if seizures should reoccur (e.g., driving or swimming). State laws defining when patients with seizure disorders are allowed to resume driving vary widely.5 Compliance to the treatment plan is sometimes difficult because of side effects of pharmacologic interventions. However, most patients are able to achieve optimal seizure control and lead active and productive lives. For some patients with seizure disorder uncontrolled by medications, surgical excision of the seizure focus may be an option. Neurostimulation is an appropriate therapy for certain patients with refractory seizures.6
Dementia
Dementia is not a specific disease but rather a syndrome associated with many pathologic processes. It is characterized by progressive deterioration and continuing decline of memory and other cognitive changes. Personality and behavior changes accompany the cognitive deterioration. Judgment, abstract thinking, and complex task performance are all affected. The onset of dementia may be insidious, and the affected individual may initially appear uninterested or lacking initiative. Many demented patients have agnosia or lack of insight into their cognitive deficiencies.
Alzheimer disease accounts for 60% to 80% of all dementias, whereas vascular dementia is the second most common cause. An estimated 1 in 8 people older than 65 have Alzheimer disease. It affects nearly half of those 85 years of age and older.7
Etiology
Multiple causes/types of dementia exist, and a full discussion of each is beyond the scope of this chapter. Some examples of dementia-causing illness include alcoholism, intracranial tumor, normal-pressure hydrocephalus, Parkinson disease, Lewy body disease, Huntington disease, multiple sclerosis, Pick disease, Creutzfeldt-Jakob disease, and bovine spongiform encephalopathy (mad cow disease). Unfortunately, these also can occur in combination, causing severe disease. Because Alzheimer- and vascular-type dementias are the first and second most common causes of dementia, they will be discussed in detail. The subsequent discussion of treatment of individuals with dementia will be more general because the care issues are similar regardless of type of dementia.
It is important to consider all potential causes of cognitive change when dealing with patients with mental status change. Both delirium and depression in the elderly can cause signs and symptoms that resemble those of dementia. Delirium is a global mental dysfunction that includes disturbed consciousness, decreased awareness of the environment, inability to maintain attention, disrupted sleep-wake cycles, drowsiness, restlessness, emotional lability, incoherence, and hallucinations.8 Symptoms of delirium tend to have an abrupt onset and may fluctuate often, becoming worse at night. Delirium can result from numerous causes such as medication/polypharmacy, metabolic abnormalities, nutritional deficiencies, and infection, among others. Delirium may occur more frequently in individuals with an underlying dementing illness.
Pathogenesis
The hallmark pathophysiologic changes associated with Alzheimer disease include intracellular neurofibrillary tangles and extracellular amyloid (senile) plaques (Figure 45-1). As a result of these changes, diffuse neuronal damage and brain atrophy occur. The brain of a patient with advanced Alzheimer disease often weighs up to 20% less than a normal brain.9 The temporoparietal and anterior frontal regions of the brain are chiefly affected, exhibiting enlarged sulci and ventricles and atrophic gyri (Figure 45-2). Neurofibrillary tangles are composed of helical filaments formed from hyperphosphorylated protein tau, also known as neural thread protein.9 In the central nervous system, neural thread proteins bind and help stabilize microtubules (the cell’s internal support structure or skeleton). Inflammatory changes, lipid abnormalities, and aging are among the processes thought to be responsible for activating the phosphorylating enzymes altering the structure of the tau proteins.10 The presence of neurofibrillary tangles is well correlated with dementia; however, neurofibrillary tangles are not specific just to Alzheimer disease and are found in other neurodegenerative disease processes.11 The second, but most specific, change in the brain of patients with Alzheimer disease is the deposition of extracellular amyloid plaques.
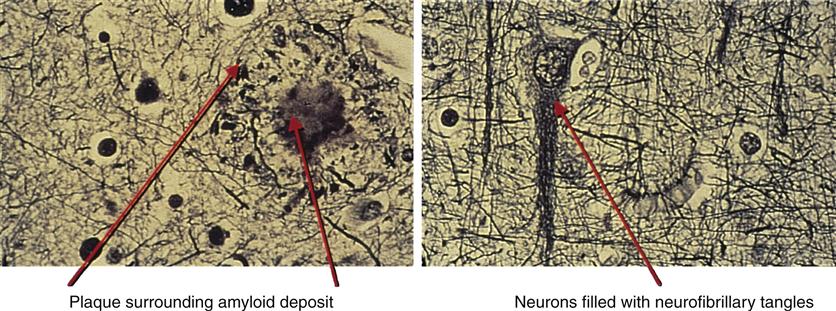
It is not known whether amyloid plaques cause Alzheimer disease or result from it. The number of senile plaques seems to correlate with the severity of disease. In plaques, β-amyloid is a protein fragment snipped from a larger protein—amyloid precursor protein (APP)—during metabolism. APP is a member of a large family of proteins that are associated with cell membranes. During metabolism, APP becomes embedded in the membrane of the nerve cell, partly inside and partly outside the cell. While APP is embedded in the cell membrane, proteases cleave APP apart. β-Amyloid is produced only when the cleavage happens at the wrong place in APP.
After β-amyloid is formed, it is not known how it moves through or around the nerve cells. In the final stages of its journey, it joins with other β-amyloid filaments and fragments of dead and dying neurons to form the dense, insoluble plaques that are a hallmark of Alzheimer disease in brain tissue. Inflammatory processes including acute-phase response, complement activation, and accumulation of activated microglia and astrocytes accompany the amyloid deposition and neurofibrillary tangle formation.10,11 The accumulation of β-amyloid also causes oxidation of lipids, activation of apoptotic genes, disruption of cell membranes, and excitotoxicity from the neurotransmitter glutamate.10
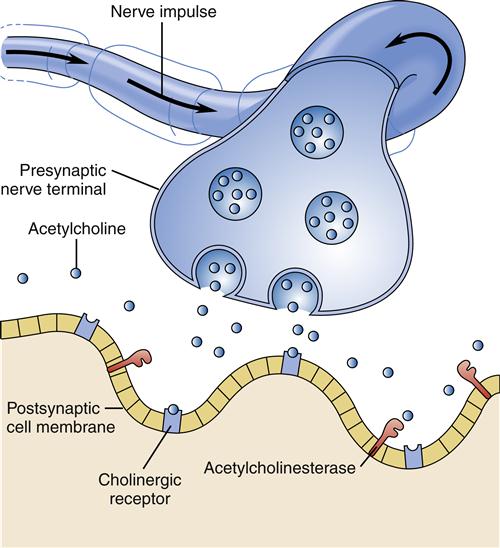
Much interest exists in the neurotransmitter systems in relation to Alzheimer disease. Damage in Alzheimer disease involves changes in three mechanisms: nerve cell communication, metabolism, and repair. Several studies have found abnormalities in the cholinergic system, including reduced activity of choline acetyltransferase (the enzyme necessary for acetylcholine synthesis) and decreased acetylcholine synthesis (Figure 45-3). Some researchers believe that β-amyloid may be responsible for lower choline levels in nerve cells and decreased acetylcholine levels. The degeneration of cells in the nucleus basalis, a band of gray matter in the ventral portion of the medulla oblongata, has also been linked to diminished levels of acetylcholine in the cerebral cortex, a finding that provides further evidence for the significant role of the cholinergic system in Alzheimer disease.9 Along with alterations in acetylcholine, other neurotransmitters are also affected. Imbalances in the activity of glutamate, dopamine, and serotonin contribute to the behavioral signs and symptoms of Alzheimer disease.12
Vascular dementia results from single cerebrovascular insults (such as cerebral infarction), from multiple lacunar infarcts, or from microvascular pathology. Microvascular insults may not show any localizing clinical symptoms and may be found incidentally on brain imaging. However, the presence of these also does not automatically mean a diagnosis of vascular dementia.13 The symptoms of vascular dementia may be similar to those of Alzheimer dementia. Most research/reports present vascular dementia and Alzheimer dementia as entirely separate entities; however, there is increasing evidence that particularly in elderly patients the brain lesions associated with both often coexist. There is emerging evidence that the cascade of events leading to the development of Alzheimer disease plaques and tangles may be due to ischemic and inflammatory insults of cerebrovascular disease.10,13
The primary risk factors for the development of Alzheimer disease include age and family history. Epidemiologic studies show that individuals who have an affected first-degree relative with Alzheimer disease have a fourfold greater risk of developing the disease. The risk is greater if there are individuals in more than one generation with the disease.7 Three main genes have been identified and are thought to be responsible for autosomal dominant familial Alzheimer disease. Presenilin 1, presenilin 2, and APP are believed to increase the amount of β-amyloid protein.11 These genes are rare, accounting for only 3% of Alzheimer disease cases, usually the early-onset variant.11 The major gene associated with late-onset Alzheimer disease is apolipoprotein e4 (APOe4). By age 85, those who are homozygous for the APOe4 allele have a 50% to 90% chance of developing Alzheimer disease. Those who are heterozygous have a 45% chance of developing the disease. Carrying the APOe4 gene has also been associated with increased risk of atherosclerosis and cerebrovascular disease, strengthening the link between Alzheimer disease and vascular dementia.10,13 Lifestyle has also been linked to the risk of Alzheimer disease. Head trauma, diabetes, and depression have been linked to an increased incidence of the disease as well as marital status, urban living, and inactive mental and physical lifestyle.13 Risk factors for vascular dementia include those for stroke, hypertension, and diabetes.13,14 Clearly, both vascular dementia and Alzheimer disease include a complex interplay of genetic, environmental, and lifestyle factors.
Clinical manifestations
Regardless of when patients first present with dementia, it is likely that brain disease has been present for quite some time. Most patients experience a gradual onset with a chronic progressive decline in cognitive functioning. There is memory loss, especially in short-term memory, whereas long-term memory may be preserved. Thinking ability declines, and there is a decreasing ability to function at work and in social settings. Anxiety and agitation are common. As the disease progresses, individuals have increasing difficulty with judgment, problem solving, and communication. Assistance may be necessary for completing activities of daily living (ADLs). Difficulty with eating and swallowing, and weight loss are common. Loss of bladder and bowel control and eventual complete loss of the ability to ambulate occur in the late stages. Accidents and infection are common causes of death.7
Diagnosis and treatment
The initial evaluation of a patient thought to have dementia of any type begins with a complete history and physical examination. This should address the patient’s overall general health and any coexisting medical conditions. All manageable causes for dementia or delirium should be ruled out. It is recommended that the evaluation should include a complete blood cell count, chemistry panel, thyroid function, vitamin B12 levels, and syphilis serology. Other testing such as Lyme serology, human immunodeficiency virus (HIV), urinalysis with culture/sensitivity, heavy-metal assays, sedimentation rate, and other vitamin levels may be warranted in certain patient situations. Other evaluations such as a chest x-ray and lumbar puncture may also be helpful. Neuroimaging may include computerized tomography (CT) and magnetic resonance imaging/magnetic resonance angiography (MRI/MRA), which may identify vascular disease, normal-pressure hydrocephalus, tumors, abscesses, or subdural hematoma. PET (positron emission tomography) scans are not routinely recommended at this point, but show promise in identifying Alzheimer disease when combined with a history of genetic risk. Mental status examinations, the clock drawing test, and tests of functional status are recommended.9,11,14 A current list of the patient’s medications, including over-the-counter medications, must also be reviewed. Medications with anticholinergic actions/side effects are a common cause of changes in cognitive functioning in the elderly.
Early diagnosis and intervention are key in the management of dementia. The financial and legal ramifications of dementia can be devastating to patients and their families and caregivers. If the diagnosis is made before the onset of severe cognitive disability, the patient can be involved in decisions regarding long-term care, power of attorney, and living will issues. Early diagnosis is also vital to initiating therapy as early as possible.
Currently, two classes of drugs are approved by the Food and Drug Administration for the treatment of Alzheimer disease. The first class is the acetylcholinesterase inhibitors: tacrine (Cognex), donepezil (Aricept), rivastigmine (Exelon), and galantamine (Reminyl). These agents are indicated for use in patients with mild to moderate Alzheimer disease. Although not a cure, the acetylcholinesterase inhibitors have been shown to stabilize cognitive function and slow progression of the illness.14,15 Acetylcholinesterase inhibitors have also been shown to improve cognitive functioning in patients with vascular dementia.15
The second class of drugs used in the treatment of Alzheimer disease is known as the N-methyl-D-aspartate (NMDA) receptor antagonists. Currently only one drug in this class is available in the United States. Memantine (Namenda) is indicated for the treatment of moderate to severe Alzheimer-type dementia. This drug blocks stimulation by the neuroexcitatory transmitter glutamate. Again, this medication is not a cure, but slows progression of the disease.16 Studies using combination therapy of acetylcholinesterase inhibitors and NMDA antagonists are showing modest improvement in cognitive functioning.15,16 Studies using NMDA antagonists for vascular dementia have not shown conclusive benefit.17
Many other medications, although not approved for use in treating Alzheimer disease, are used to manage the symptoms, such as depression, sleep disturbance, agitation, and psychosis. These medications include antidepressants, anxiolytics, antipsychotics, and mood stabilizers.16
A variety of medications and nutritional supplements have been used in the prevention and management of Alzheimer disease, including gingko biloba; antioxidants such as vitamin E, α-lipoic acid, omega-3 fatty acids, and coenzyme Q10; and nonsteroidal antiinflammatory drugs (NSAIDs). The blood pressure drugs angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) are showing slowed progression of disease beyond just lowering blood pressure. However, all the research has been completed on hypertensive individuals.18–20 Research findings regarding nutritional supplements are inconsistent and therefore ongoing.18
Other treatments for dementia include optimal management of other coexisting illnesses, interventions aimed at wellness, regulation of optimal nutritional intake, and protection from injury. In early stages of the disease, most patients are cared for at home, often by family members. It is important that the home environment be safe and that there be measures in place to control wandering. Consistent routines and familiar surroundings allow the patient to feel more comfortable and experience less confusion. As the disease progresses, the individual with dementia may have to be placed in an alternative living situation such as a nursing home or assisted-living program. Caring for the caregivers of patients with dementia is important.
Parkinson Disease
Parkinson disease is a disorder of mobility that affects 1 million Americans. It is estimated that 60,000 new cases are diagnosed each year. Although it usually develops after age 65, 4% of those diagnosed are younger than age 50.21
Etiology
Parkinson disease may be idiopathic or acquired. Idiopathic Parkinson disease is that in which no demonstrable cause is identified. Common causes of acquired parkinsonism include infection, intoxication, and trauma.3 Typically, parkinsonism attributable to drug toxicity evolves rapidly, unlike the slow, insidious onset of the idiopathic form of the disease. Side effects of drugs of the phenothiazine class (e.g., chlorpromazine, prochlorperazine, and thioridazine) and butyrophenone class (e.g., haloperidol) may manifest in a parkinsonian syndrome at toxic levels. Discontinuing the medication generally results in improvement in the symptoms. However, the additional use of the anticholinergic antiparkinsonian drugs may aid in more rapid recovery. The rest of this discussion of Parkinson disease refers to the most common idiopathic type.
Pathogenesis
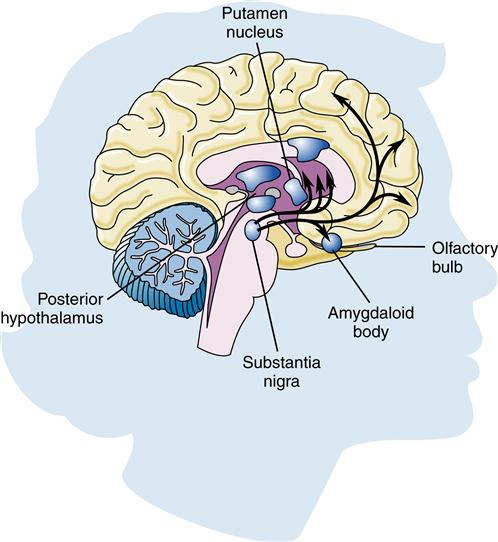
Parkinson disease results from degeneration of the pigmented dopaminergic neurons found in the substantia nigra (Figure 45-4) and, to a lesser extent, neurons elsewhere in the brain. Eosinophilic cytoplasmic inclusions known as Lewy bodies may be found in the surviving neurons. Incidentally, Lewy bodies are found along with amyloid plaques at autopsy in the brains of some patients with a severe form of dementia. This suggests a possible link with Alzheimer disease. The exact cause of this degeneration is unknown, but mitochondrial dysfunction from oxidative stress, genetics, and environmental toxins has been implicated.22 Adaptive immunity may play a part in the progression of Parkinson disease by reacting to the abnormal proteins and causing neuroinflammation.23
At least 13 different gene groups have been identified as having a role in the development of Parkinson disease. In particular, identification of a mutation in the α-synuclein gene has become the focus of much interest. Although mutations of the gene are a rare cause of Parkinson disease, α-synuclein is abundant in neurons, specifically in presynaptic terminals, and is a major component of Lewy bodies.22 Another gene identified in the development of Parkinson disease is parkin. Mitochondrial quality control is mediated by parkin. When functioning normally, it selectively recognizes and eliminates damaged mitochondria from the cell by autophagy. Parkin mutations are associated with monogenic forms of Parkinson disease.24 Although genes have received much attention in Parkinson disease research, environmental factors have also been studied. High caffeine intake has been found to have an inverse relationship to the risk of developing the disorder.22,25 Long-term exposure to the pesticide rotenone and the herbicide paraquat have been linked to increasing risk for Parkinson disease.26 Whatever the cause of the degeneration of the dopaminergic cells, 75% to 80% of the neurons have died before any symptoms of the disease become apparent.
Clinical manifestations and treatment
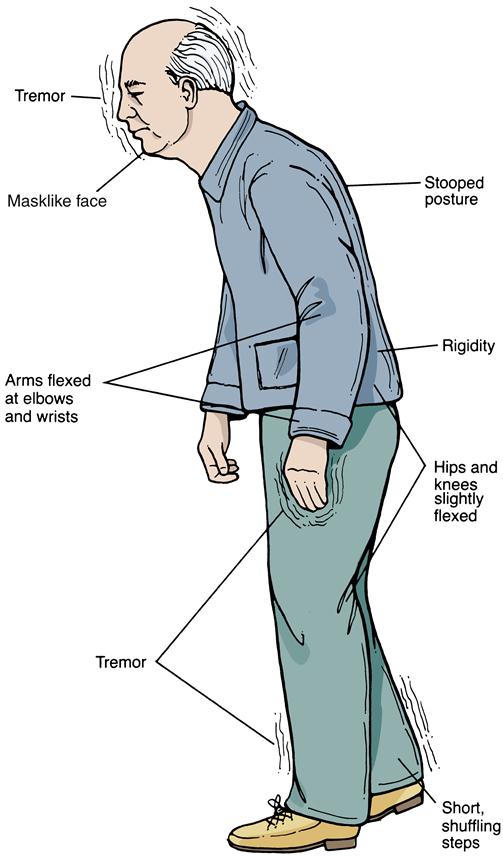
Because of the insidious onset, earlier evidence of Parkinson disease may be discovered in a thorough health history. Frequently, the very early signs of the disorder (loss of flexibility, aching, and fatigue) are overlooked by the patient or are attributed to the aging process. Initially, symptoms are usually worse on one side of the body and then progress to involve both sides. Tremor is often the first symptom recognized that prompts patients to seek treatment. The tremor is generally at rest, unilaterally affecting distal extremities. Hand tremors may be described as pill-rolling movements. Attempts to passively move the extremities are met with cogwheel rigidity. As Parkinson disease progresses, the tremor will often become bilateral/more generalized. Additional early signs of the disease include bradykinesia, rigidity, hypokinesia, loss of facial expression, and infrequent eye blinking (Figure 45-5). Again, these symptoms may be overlooked by patients but are usually apparent to observant family members.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


