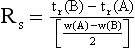
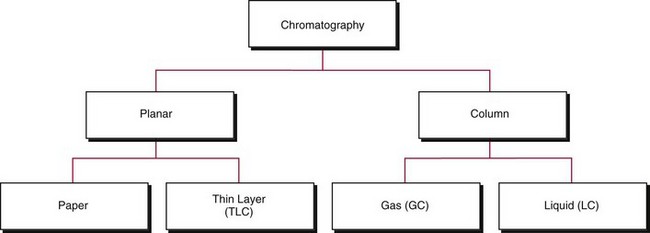
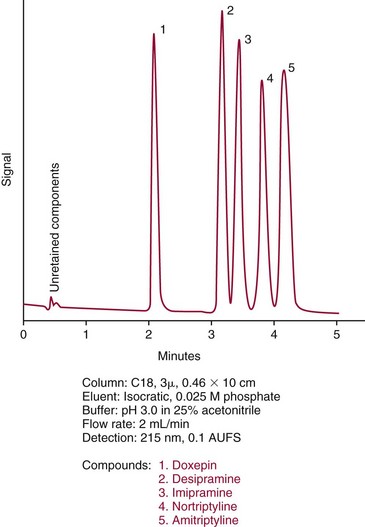
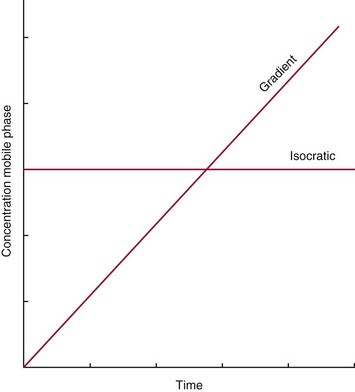
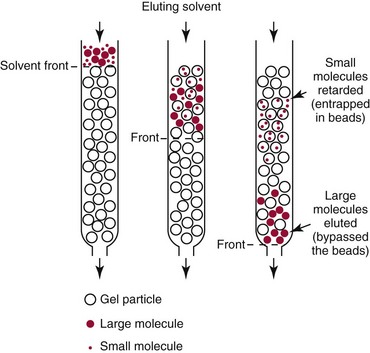
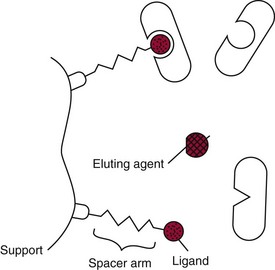
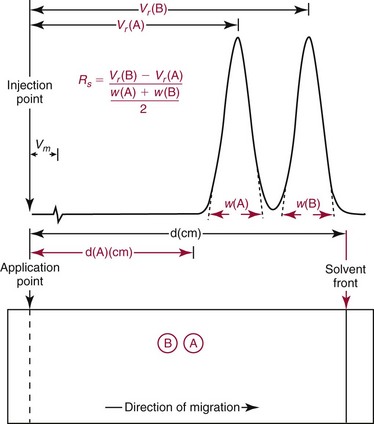
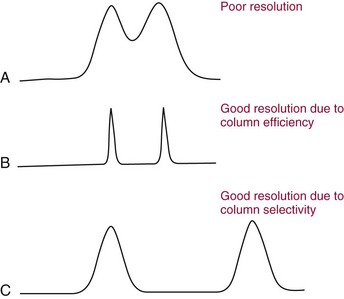
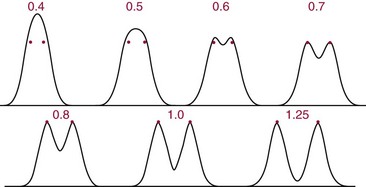
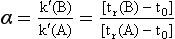Chapter 13 Biological fluids are complex mixtures, and clinical tests for specific components contained in these fluids often entail one or more sequential separation steps to isolate or enrich the component of interest. Common methods for achieving separation include (1) chromatography, (2) extraction, (3) differential precipitation, and (4) electrophoresis (see Chapter 12). This chapter describes basic concepts and principles of chromatographic and extraction techniques that are used as analytical techniques or as methods to prepare specimens for analysis. Chromatography is a process in which components of a mixture are separated by differential distribution between a mobile phase and a stationary phase.12,48,66 Components with greater distribution into the stationary phase are retained and move through the system more slowly. The initial description of this technique by Mikhail Tswett in 1903 entailed the separation of plant pigments into separate colored bands on a column of calcium carbonate.13 He originated the term chromatography, derived from chroma and graphein (Greek for “color” and “to write”), and this term has continued to be used even though most separations do not involve colored components. Planar and column are the two basic forms of chromatography (Figure 13-1). In planar chromatography, the stationary phase is coated on a sheet of paper (paper chromatography) or is bound to a solid surface [thin-layer chromatography (TLC)]. For paper chromatography, the stationary phase is a layer of water or a polar solvent coated onto paper fibers. In TLC, a thin layer of particles of a material such as silica gel is spread uniformly on a glass plate or a plastic or aluminum sheet. When the thin layer consists of particles with small diameters (4.5 µm), the technique is known as high-performance, thin-layer chromatography (HPTLC). In column chromatography, the stationary phase may be a pure silica or polymer, or it may be coated onto, or chemically bonded to, support particles.6 The stationary phase may be “packed” into a tube, or it may be coated onto the inner surface of the tube. Column chromatography includes both gas chromatography (GC)47 and liquid chromatography (LC)62; with classification dependent on whether the mobile phase is a gas or a liquid. Operationally, the instrument used to perform a GC or LC separation is known as a gas or liquid chromatograph. When the stationary phase in LC consists of small-diameter particles, the technique is known as high-performance liquid chromatography (HPLC). When a gas or liquid chromatograph is connected to a mass spectrometer, the combined or “hyphenated” techniques are gas chromatography–mass spectrometry (GC-MS) and liquid chromatography–mass spectrometry (LC-MS).23 In analytical GC and LC, the mobile phase, or eluent, exits from the column and passes through a detector that produces an electronic signal that is plotted as a function of (1) time, (2) distance, or (3) volume. The resulting graphical display is a chromatogram (Figure 13-2). Chromatographic separations are classified by the chemical or physical mechanisms used to separate the solutes (Figure 13-3). These include (1) ion-exchange, (2) adsorption, (3) partition, (4) affinity, and (5) size-exclusion mechanisms. Predominantly, clinical applications use chromatographic separations based on ion-exchange and partition mechanisms. Figure 13-3 Examples of separation mechanisms used in chromatography. (Courtesy James K. Hardy, Akron, Ohio [http://ull.chemistry.uakron.edu].) Ion-exchange chromatography relies on the exchange of ions between charged groups bound to a stationary phase and ions of opposite charge in the mobile phase (see Figure 13-3). Depending on the charge of the stationary phase, components binding to the column may be cations (positively charged) or anions (negatively charged). Cation-exchange solid phases contain covalently bound, negatively charged functional groups. Examples include strongly acidic groups, such as sulfonate ions, or weakly acidic groups, such as carboxyl or carboxymethyl. Anion-exchange solid phases have strongly basic quaternary amines, such as triethylaminoethyl groups, or weakly basic groups, such as aminoethyl (AE) and diethylaminoethyl (DEAE) groups. Separations are performed under conditions where analytes are charged and bind to the ion-exchange stationary phase. Solutes are eluted with a solution containing competing counterions, such as sodium for cation exchange and chloride for anion exchange, and, in some cases, by adjustment of pH to decrease the charge of bound ions or the ion-exchange surface. Important factors in performance of ion-exchange chromatography include (1) type of stationary phase ionic group, (2) charge density, (3) stationary phase matrix, (4) type and concentration of ions in the mobile phase, and (5) mobile phase pH. Many stationary phases exhibit mixed mode retention by a combination of ion exchange and adsorptive binding. As an example, ion-exchange resins used for amino acid analysis are able to separate amino acids with virtually the same charge because of variable adsorptive interactions of different amino acids with the stationary phase. Some solid-phase extraction media are designed to have mixed hydrophobic–ion-exchange retention, offering improved selectivity versus extraction media designed predominantly for hydrophobic or ion-exchange extractions. The basis of separation by adsorption chromatography is the difference between adsorption and desorption of solutes at the surface of a solid particle (see Figure 13-3). Electrostatic, hydrogen-bonding, and dispersive (van der Waals) interactions are the physical forces that control this type of chromatography.17,28 As solvent molecules compete for binding to the stationary phase, the strength of binding to the stationary phase depends on the properties of the stationary phase and of the mobile phase. In general, for a polar stationary phase, elution tracks the polarity of the components in a mixture: less polar ones elute first, followed by those of increasing polarity.7 Hydrophilic interaction liquid chromatography (HILIC) is a type of adsorption chromatography that is used to separate highly polar compounds that form hydrogen bonds with a stationary phase having a relatively polar surface.8,26,41 Examples of stationary phases used for HILIC include (1) unmodified silica, (2) silica with bonded zwitterionic groups, (3) hydroxyl-rich compounds, and (4) amide-rich compounds. Initial binding of solutes to the stationary phase is performed in relatively hydrophobic solvent, and elution is attained with solvents of increasing polarity. A limitation of HILIC for clinical laboratory use is that biological fluids are highly polar and include a substantial quantity of salts that also interact with polar stationary phases. Therefore, the specimen may need to be extracted or modified by the addition of a less polar solvent such as acetonitrile to promote binding to the stationary phase. Interest is growing in the application of HILIC as a separation technique linked to mass spectrometry, but most separations in clinical laboratories have relied on reversed-phase or ion-exchange separations. The differential distribution of solutes between two immiscible liquids is the basis for separation by partition chromatography (see Figure 13-3).46,55,56 Operationally, one of the immiscible liquids serves as the stationary phase. To prepare this phase, a thin film of liquid is adsorbed or chemically bonded onto the surface of support particles or onto the inner wall of a capillary column. In reversed-phase, partition chromatography, the (1) stationary phase is nonpolar, (2) mobile phase is relatively polar, and (3) hydrophobic molecules are preferentially retained. It is the most widely used technique for solid-phase extraction and liquid chromatography in the clinical laboratory. An example of separation of several antidepressant drugs by reversed-phase chromatography is shown in Figure 13-2. Examples of stationary phases for reversed-phase adsorption include silica with bonded hydrophobic groups such as octadecyl (C18), octyl (C8), phenyl, or butyl (C4) and polystyrene-divinylbenzene. For solid-phase extractions, similar materials are used including copolymers of styrene with more hydrophilic monomers that form stationary phases with balanced hydrophobic-hydrophilic character or with charged groups that offer mixed-mode extraction with both hydrophobic and ion-exchange retention.6,20,55 The retention characteristics of silica-based stationary phases depend on (1) the nature of bonded phases, (2) the amount of bonded phase (often expressed as % carbon load), (3) the surface area of the particles, (4) the pore size of the particles, and (5) the quantity of accessible silanol groups on silica particles that depend on surface treatments such as “end-capping,” where such groups are replaced by trimethylsilyl groups. For reversed-phase separations, solutions are applied in relatively polar solvent such as water or water with a low concentration of organic solvent such as methanol or acetonitrile. Reversed-phase separations are often used with aqueous specimens such as blood and urine, because most of the solvent (water) and inorganic salts pass through the column with minimal retention. The partitioning of solutes in reversed-phase separations usually is favored by adjustment of pH to minimize the charge of solutes, such as decreasing the pH so that organic acids are in uncharged forms, or adding ion-pairing agents that yield less polar ion pairs. Prior to the introduction of a sample, hydrophobic stationary phases may require conditioning or “wetting” with a solvent such as methanol before equilibrating with a more polar solvent. Some stationary phases, such as C18-silica, may undergo “phase collapse” of the bonded phase if run in completely aqueous solvent, which probably represents the aliphatic bonded phase folding down onto the surface and having decreased access to solvent. During a solid-phase extraction, drawing air into the column can similarly require reapplication of organic solvent to wet surfaces. Use of balanced hydrophobic-hydrophilic phases for extractions reduces the need for wetting of the stationary phase and avoids problems of phase collapse with loading of aqueous solutions.19,55 Polymeric stationary phases usually have higher capacity than bonded silica stationary phases, because the entire particle serves as the stationary phase. More hydrophobic solvents generally are stronger eluents for reversed-phase separations. For normal-phase chromatography (polar stationary phase, nonpolar mobile phase), the eluotropic series is reversed. In practice, it is also possible to change the solvent strength by using mixtures of solvents and gradients of changing proportions of solvents during an analysis (Figure 13-4).9 It is also possible to modify the polarity of aqueous solutions by changing the salt concentration. This characteristic is employed in a variation of reversed-phase chromatography termed hydrophobic interaction chromatography, which has been applied mainly for separation of proteins. Binding to a weakly hydrophobic stationary phase containing interaction groups such as phenyl or butyl groups is promoted by loading specimens in a high-salt concentration, and elution is achieved by lowering the salt concentration rather than by adding an organic solvent. A wide range of solid phases for reversed-phase separations have slightly different specificities, and some have mixed modes of separation.6,16,39 Some solid phases such as porous graphite or fluorinated hydrocarbons or hydrophobic stationary phases with embedded polar groups offer different selectivities than traditional C18-silica. Stationary phases bonded on silica have weak cation-exchange and normal-phase properties, depending on the quantity of accessible hydroxyl groups on the silica. Basic compounds may exhibit peak tailing caused by ion exchange with the silica. Ion-exchange properties of silica are minimized by chemically modifying the surface to block hydroxyls and by operating at low pH. Silica tends to dissolve slowly at pH > 7, so that silica particles usually require separations below pH 8, unless the silica is stabilized by surface treatment.5 Other types of stationary phases such as polystyrene or porous graphite are stable at high pH and allow separations over a broader pH range. Because of its wide diversity of stationary phases with differing selectivities and varying ability to change solvent conditions and ion-pairing agents, reversed-phase chromatography is a very versatile technique, and separations often achieve high efficiency and, sharp peaks. Compounds representing the greatest challenge for reversed-phase separations are small ions and highly polar compounds such as sugars or amino acids, which tend to be weakly retained by reversed-phase media, and basic compounds, which may exhibit peak tailing as the result of interactions with silica. Derivatization of some compounds such as amino acids has been employed to improve reversed-phase retention and to add chromophores or fluorophores to improve detection.48 Separation of large peptides or proteins requires stationary phases with larger pore sizes than are routinely used for separation of small molecules.3,32,63 Many biological molecules occur as specific stereoisomeric forms, such as L-amino acids, and in some cases, only a particular stereoisomer of a therapeutic drug may be active. In general, the two mirror-image forms of a compound or enantiomers have very similar physical properties and do not separate in most chromatographic analyses. Separation of enantiomers generally requires use of a stationary phase that has an enantiomeric component that will interact with molecules in a stereospecific manner. For example, chiral stationary phases (CSPs) that contain groups such as derivatized L-amino acids or stereospecific carbohydrates have been used to separate enantiomers.52,62A,72 Size exclusion chromatography, also known as (1) gel filtration, (2) gel permeation, (3) steric exclusion, (4) molecular exclusion, or (5) molecular sieve chromatography, is a technique that separates molecules or other particles based on size (Figure 13-5). In this technique, the stationary phase consists of porous particles with an inert surface designed to have minimal adsorption of components. Materials used for stationary phases include cross-linked dextran, polyacrylamide, agarose, polystyrene, and porous glass with an inert surface coating. Beads of these materials are porous and have pore sizes that allow small molecules to enter. Molecules too large to enter the pores remain entirely in the mobile phase and are rapidly eluted from the column. Molecules that are intermediate in size have access to various fractions of the pore volume and elute between large and small molecules according to the following relation: By calibrating a size-exclusion column with molecules of known size, it is possible to estimate the molecular size of proteins or other molecules.64 However, this is a relatively low-resolution technique, and separation of components requires substantial proportional differences in molecular weight. The diameter of globular proteins changes only in proportion to the cube root of molecular weight, so that an eightfold difference in molecular weight is required to yield a twofold change in diameter.64 Linear polymers, such as DNA, and proteins denatured with sodium dodecylsulfate or guanidine hydrochloride have a much larger diameter for the same molecular weight, and diameter changes approximately in proportion to the square root of molecular weight, allowing better resolution of linear molecules relative to molecular weight than of globular molecules. Therefore, analysis of molecules under denaturing conditions provides the ability to resolve molecules with smaller differences in molecular weight. Specific molecular interactions are used as the basis for affinity chromatography (Figure 13-6).25,57 Examples include (1) interactions of antibody with antigen, (2) enzyme with substrate, (3) aptamer with ligand, (4) receptor with ligand, and (5) lectin with sugar. The stationary phase for affinity chromatography is prepared by immobilizing one component of an interaction pair and using it to capture the other component of the interaction pair. Orientation of the capture molecule and its accessibility are important elements in the function of capture molecules. Using a spacer to provide distance from the surface of the stationary phase may improve access and function of the capture molecule. An alternative approach to developing affinity matrices without immobilized ligands is to form molecularly imprinted surfaces on the capture phase.38 The shape and structure of pores formed by molecular imprinting yield selective binding of molecules that have a complementary structure. The power of affinity chromatography lies in its selectivity. Affinity chromatography is widely applied to purifying antibodies and other proteins used in clinical assays. Lectin affinity chromatography offers a means of assessing changes in the oligosaccharide structures bound to glycoproteins. Immobilized phenylboronic acid has been used to separate glycated hemoglobin from unmodified hemoglobin. Most heterogenous assays in the clinical laboratory are based on the selective extraction or capture of a specific target molecule onto a solid phase with elution of all other molecules (see Chapter 16). Immunodepletion or immunopartitioning of high-abundance proteins has been used as a means of preparing biological specimens for proteomic analysis to assist in the detection of lower-abundance components.53 Resolution (Rs) is a measure of the separation between two components (Figure 13-7). Improved resolution may result from sharper peaks (higher chromatographic efficiency) or from a greater separation between peaks (higher selectivity), as described in Figure 13-8. It is expressed mathematically as follows: tr(A) = retention time for solute A tr(B) = retention time for solute B w(A) = peak width (units of time) measured at base for solute A w(B) = peak width (units of time) measured at base for solute B Retention may be expressed in units of time (previous equation) or volume (see Figure 13-7). A resolution of 1.25 or greater reflects baseline separation of peaks; lower resolution represents increasing degrees of overlap between peaks, as described in Figure 13-9. Resolution is improved by enhancing efficiency (achieving narrower peaks) or by improving selectivity to increase the separation between the two components. Two ways to increase resolution are by using a longer column and by selecting a column with higher efficiency. Either of these measures increases N, the number of theoretical plates. Resolution increases proportionally with the square root of N, so that doubling of column length (doubling of N) will increase resolution by a factor of 1.41. Therefore, the greater the solute affinity for the stationary phase, the smaller the Rf value. A = a constant related to eddy-diffusion B = a constant related to longitudinal diffusion C = a constant related to kinetics of mass transfer between mobile and stationary phases Factors that affect chromatographic efficiency are listed in Box 13-1. In practice, efficiency is improved by using (1) smaller particles, (2) nonporous particles, or (3) a coated capillary surface as the stationary phase to minimize mass transfer effects (the C constant in the van Deemter equation). This allows the use of higher mobile phase flow rates without peak broadening, resulting in faster separations. The limiting factor in reducing particle size is the increased resistance to flow and the need to operate at higher pressures. Peak capacity is the theoretical maximum of the number of components that are able to be separated in a single chromatographic analysis20,51,54; it assumes a continuous distribution of peaks separated by four standard deviations. In practice, the number of components that are resolved will be lower than expected, because retention times of components are not evenly distributed. The peak capacity, therefore, serves as an upper limit for the number of components that are able to be resolved and as a performance measure for a chromatographic method. The peak capacity of high-performance liquid chromatographic (HPLC) separation usually is limited to several hundred peaks. Some gains in peak capacity are achieved by (1) increasing column length, (2) elution with solvent gradients, (3) extended run times, or (4) increasing chromatographic efficiency. However, the greatest gain in peak capacity is achieved by using two-dimensional or multidimensional chromatography, where the different dimensions usually are performed sequentially (with the second dimension representing analyses of fractions collected from the first dimension).20,58 Peak capacities of greater than 10,000 have been achieved for two-dimensional HPLC separations, but this requires prolonged analysis time for each sample to perform multiple runs in the second dimension. Linking of chromatographic methods to other analytical methods or detectors, such as mass spectrometers, with the capacity to resolve or detect multiple components serves as an alternative approach to overcome the fundamental limitation of chromatographic peak capacity without extending analysis time. This enables the practical analysis of up to thousands of components in a single specimen, such as for metabolomic22 or proteomic analysis.42
Chromatography and Extraction
Chomatography
Separation Mechanisms
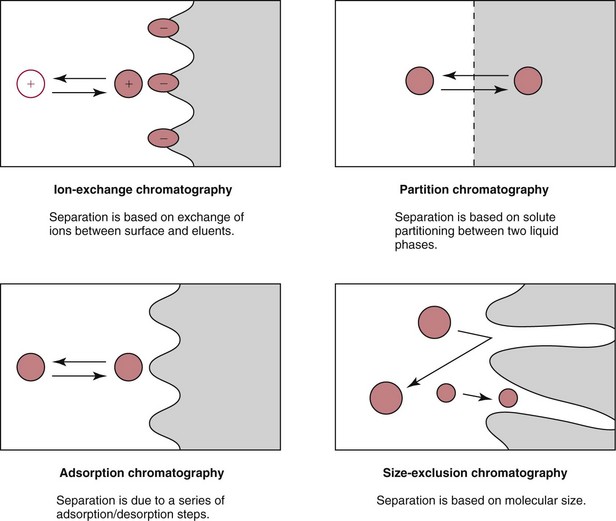
Ion-Exchange
Adsorption
Partition Chromatography
Chiral Separations
Size Exclusion Chromatography
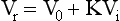
Affinity Chromatography
Resolution
Retention Factor
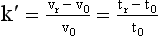

Efficiency (N)

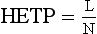
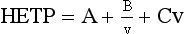
Peak Capacity
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Chromatography and Extraction
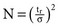 , where tr is retention time and σ is the standard deviation of the curve measured as time. A practical way to calculate N is based on the measurement of peak width (
, where tr is retention time and σ is the standard deviation of the curve measured as time. A practical way to calculate N is based on the measurement of peak width (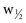 ) at half the maximal peak height;
) at half the maximal peak height; 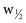 represents 2.354 σ. Therefore,
represents 2.354 σ. Therefore,