Fig. 12.1
Histologic features of normal choroid plexus and choroid plexus tumors. (a) Normal choroid plexus demonstrating typical “cobblestone” appearance (40×). (b) Choroid plexus papilloma, in contrast to normal choroid plexus in 2A, displays more elongated/columnar cells crowded in a pseudostratified manner and a smooth rather than cobblestone appearance (40×). (c) Atypical choroid plexus papilloma, with increased cellularity and focal blurring of the papillary architecture (20×). (d) Choroid plexus carcinoma showing focal papillary architecture (arrow) adjacent to an area where the papillary architecture is more blurred (40×).
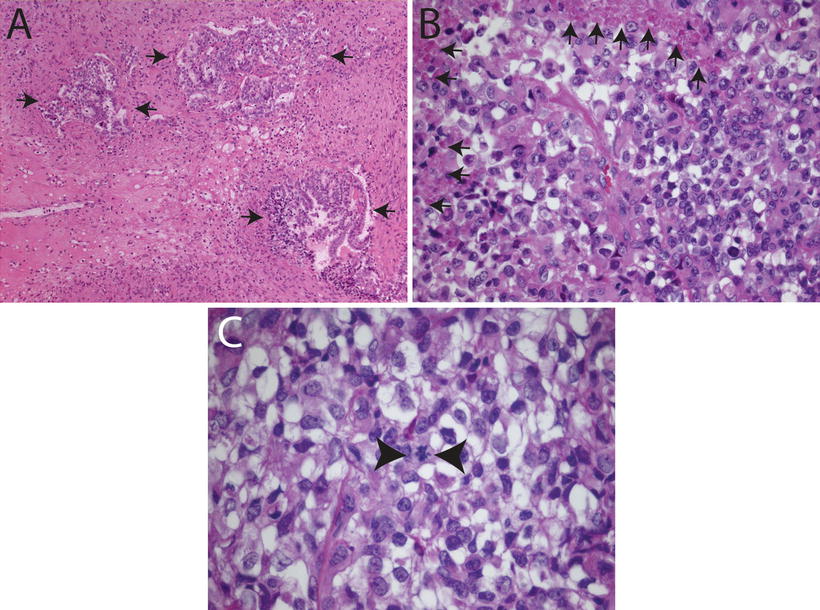
Fig. 12.2
Histologic features of choroid plexus carcinomas. (a) Choroid plexus carcinoma invading surrounding brain tissue (arrows, 10×). (b) Choroid plexus carcinoma exhibiting increased cellularity, nuclear pleomorphism, blurring of papillary architecture, and focal areas of necrosis (arrows, 40×). (c) Choroid plexus carcinoma showing loss of papillary architecture, increased cellularity, nuclear pleomorphism, and a mitotic figure between arrowheads (63×).
Clinical Features
Epidemiology
CPTs are rare, representing between 4 and 9 % of brain tumors in population-based studies [3, 4]. The typical age of presentation is the first year of life. However, CPTs can occur as rare congenital brain tumors [5, 6] and in adults, including the elderly [7–9]. These tumors also occur in other species including canines [10–14]. While most tumors are sporadic, CPTs are reported in: Li–Fraumeni syndrome (discussed below), pediatric patients with large melanocytic skin lesions [15], Pierpont syndrome [16], hypermelanosis Ito [17], and Aicardi syndrome [18]. CPPs can also be seen in von Hippel–Lindau disease [19], but it is not known if loss of the VHL allele contributes to the pathogenesis of these tumors. It is possible that these abnormalities indicate rare pathways of tumorigenesis that are yet to be elucidated.
Clinical Presenting Features
Raised intracranial pressure by hydrocephalus is the main presenting feature of CPTs [20, 21]. Other presenting symptoms may include blindness, non-focal symptoms such as convulsions [20] or focal signs such as hemiparesis [22, 23]. Large amounts of cerebral spinal fluid (CSF) production may also be encountered even when a drain or shunt is placed [24]. Sometimes highly vascularized tumors present as acute intracranial hemorrhage without previous symptoms [25]. Tumor locations in the cerebello-pontine angle may result in more specific signs such as unilateral rhinorrhea and otorrhea [26], or trigeminal neuralgia [27].
Imaging and Tumor Localization
The typical imaging characteristics of CPTs include intraventricular location and contrast enhancement indicating a high degree of vascularization. Invasion of brain tissue is a characteristic finding for CPCs as opposed to CPPs. CPTs are more common in the lateral ventricles [28] than in the third or fourth ventricles [29]. The cerebello-pontine angle is a typical location when the tumor originates in the choroid plexus of the fourth ventricle [30]. Ectopic locations unrelated to the ventricles/choroid plexus have also been described [23, 31–34]. Lateral ventricle tumors are more frequent in infants, while cerebello-pontine angle tumors occur more frequently in adults [30]. An unusual feature of CPTs is that even WHO grade I and II tumors have the potential to metastasize [35–39]. The typical metastatic route is through the CSF [39, 40], often to the spine but also with surprising frequency to the internal ear canals, pituitary stalk, and interpeduncular fossa [41]. Metastases can occur sometimes many years after primary tumor resection. Imaging of the entire craniospinal axis is recommended [40, 42, 43]. Rarely, hematogeneous metastases can occur to other parts of the brain [44], abdomen [45], bone [46, 47], and lung [48]. Abdominal seeding has been described in patients with ventricular-peritoneal shunts [49].
Imaging techniques for CPTs differ in several aspects from that for other brain tumors. As CPTs are the most frequent brain tumors detected on prenatal ultrasound [50–53], ultrasound remains an important diagnostic tool until the fontanels close [6, 51, 54]. On magnetic resonance imaging (MRI), CPTs show inhomogeneity on T2-weighted images, and moderate to marked contrast enhancement. Diagnostic specificity increases when age and intraventricular location are considered. Contrast enhancement, a common finding in these tumors, might be related to the rich vascular stroma. However, extraventricular tumors might not demonstrate contrast enhancement [34]. Differentiating the degree of malignancy on MRI can be difficult. Nevertheless, some general patterns may be observed. CPPs are usually irregular, lobulated, and solid-cystic masses, whereas CPC may present as a poorly defined, mixed-intensity mass [55]. Extensive peritumoral edema and necrosis is more frequent in CPC than in CPP [56, 57]. A thin capsule may be seen in CPP [55].
More recently, nuclear medicine methods and molecular imaging may improve diagnostic ability and also address challenging clinical issues including how to distinguish between tumor and postsurgical scars/postradiation pseudo-progression. For example, sestamibi, an agent that accumulates in mitochondria, has been used to distinguish CPTs from other brain tumors or postsurgical scars [58–62].
Pathology and Diagnostics
Macroscopy
CPTs appear macroscopically as space-occupying lesions located in the ventricles and, less commonly, in extraventricular locations. Grossly, CPPs are well demarcated with a cauliflower-like appearance. They may be attached to the ventricular wall. CPCs usually show varying degrees of invasion into the surrounding brain. High vascularity and hemorrhages are frequent.
Histopathology
CPTs are typically comprised of epithelial cells with a round or oval nucleus and small amount of surrounding cytoplasm. CPTs are fragile, and drop metastases due to CSF spread are not uncommon [63]. Key cytologic features of CPTs in the CSF include variably sized clusters to frank papillary fragments, and cells that retain epithelial features such as sharply defined cell borders [43, 64].
Specific features of the different CPTs are described below, but from a practical point of view, these tumors can be histologically classified by where they fall along the spectrum of three key histologic features: (1) growth pattern—papillary to solid, (2) mitotic activity/cellular atypia—few or none/absent to moderate/moderate to severe, (3) necrosis—little to none or prominent.
CPPs, the least malignant of CPTs, are composed of delicate fibrovascular connective tissue fronds covered by a single layer of epithelial cells with round or oval monomorphic nuclei (Fig. 12.1b). CPPs can be distinguished from normal choroid plexus (Fig. 12.1a) by an overall increased amount of choroid plexus epithelium with flatter papillae (compared to the typical cobbled stone appearance of normal choroid plexus) comprising cells with increased nuclear to cytoplasmic ratios (Fig. 12.1a, b). These cells rest upon a basement membrane that can be elucidated with special stains for collagen. Mitotic activity is extremely low. Brain invasion, high cellularity, necrosis, nuclear pleomorphism, and focal blurring of the papillary pattern are unusual, but may occur and should prompt the consideration of APP. Rarely, CPPs acquire unusual histological features, including oncocytic change, mucinous degeneration, melanization as well as formation of bone, cartilage, adipose tissue, or neuropil islands. CSF-mediated metastases can occur despite the histologic classification of WHO grade I [38].
An intermediate grade CPT, the atypical choroid plexus papilloma (APP) is recognized by the WHO (Grade II) (Figs. 12.1c and 12.3b). These tumors are defined by the presence of two or more mitoses per ten HPFs in what otherwise appears to be a CPP [1, 2]. Additional histologic features that have been reported in APP by some authors include increased cellularity, nuclear pleomorphism, solid growth, and necrosis. However, none of these features are required for the diagnosis of APP and the isolated occurrence of atypical histological features does not automatically imply malignancy.
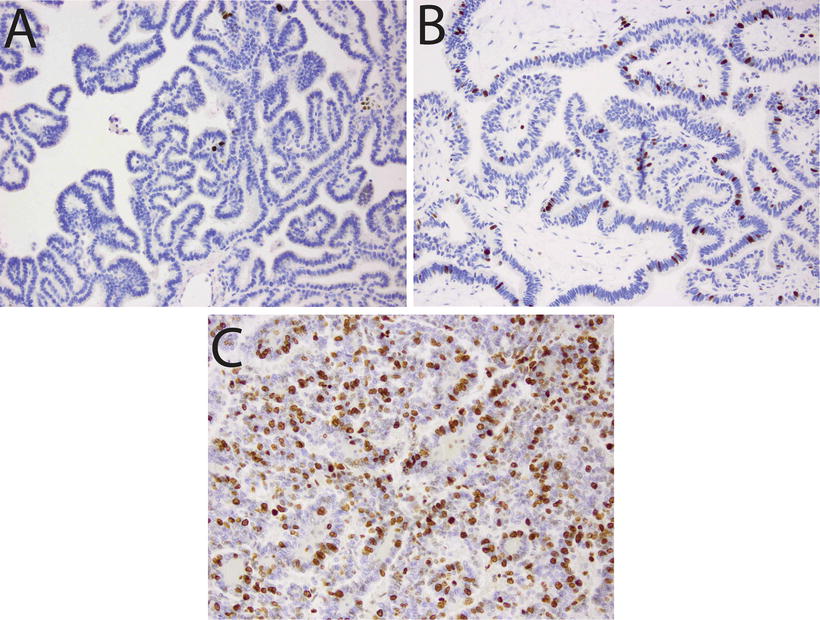
Fig. 12.3
Proliferative activity in choroid plexus tumors. Immunostains for Ki67/MIB1 illustrating: (a) Choroid plexus papilloma with low mitotic index (20×). (b) Atypical choroid plexus papilloma, with intermediate proliferative activity (20×). (c) Choroid plexus carcinoma, showing high Ki67/MIB1 labeling (20×).
CPC is a high-grade (WHO III) tumor of the choroid plexus that demonstrates frank signs of malignancy. In contrast to lower grade CPTs, CPCs demonstrate at least four of the following five features (1) frequent mitoses (Fig. 12.2C, typically more than 5 per 10 HPF), (2) increased cellular density (Figs. 12.1d and 12.2 b, c), (3) nuclear pleomorphism (Figs. 12.1d and 12.2b, c), (4) blurring of the papillary pattern (Figs. 12.1d and 12.2b, c), and (5) necrosis (Fig. 12.2b). Invasion of adjacent brain tissue by CPC is common (Fig. 12.2a). In CPCs that are truly anaplastic, identification of epithelial features may become quite challenging.
Immunohistochemistry
CPTs demonstrate expression of a wide range of immunohistochemical markers, a reminder that despite their epithelial appearance, these cells have a neuroepithelial developmental origin. The typical, though variably expressed, pattern includes S-100 protein, synaptophysin, vimentin, cytokeratins (Fig. 12.4d), glial fibrillary acidic protein (GFAP) (Fig. 12.4c), and transthyretin (TTR) (Fig. 12.4a) [65–67]. While TTR is expressed in normal choroid plexus and in many CPTs, it is unfortunately nonspecific and therefore unreliable as a precise marker of choroid plexus origins in any given tumor. However, immunohistochemical detection for membranous expression of the inward rectifier potassium channel Kir7.1 is considered specific for CPT [68] (Fig. 12.5). The Ki67/MIB index can be helpful in refining tumor grade [69] (Fig. 12.3).
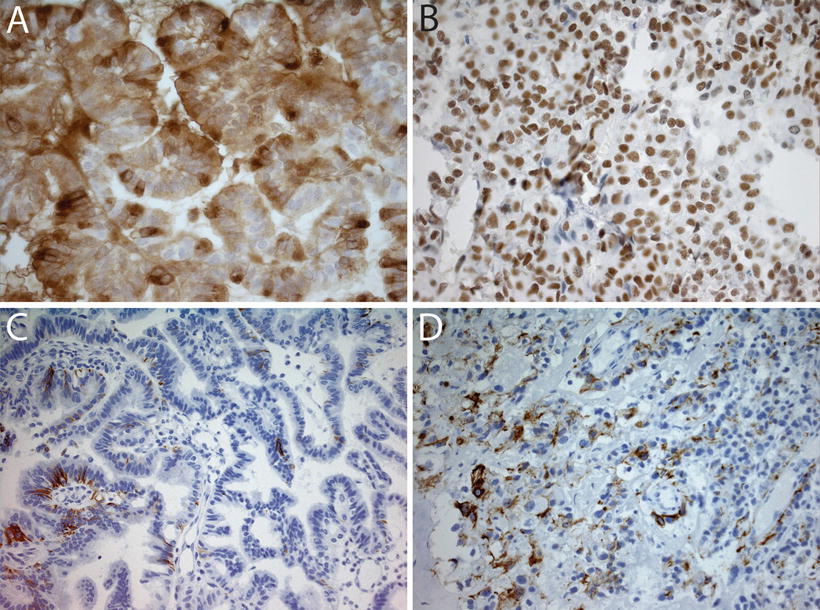
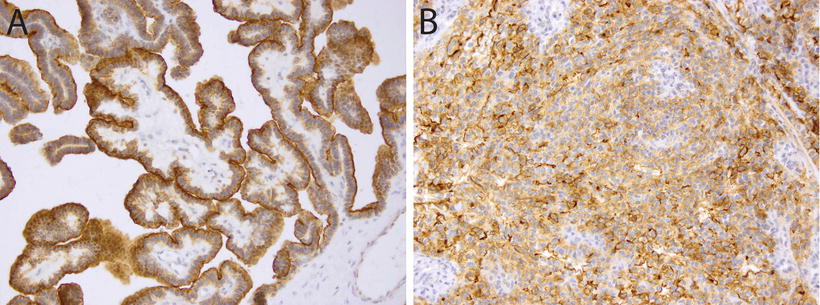
Fig. 12.4
Immunohistochemical analyses in choroid plexus tumors. (a) Immunostain for transthyretin in a choroid plexus papilloma showing diffuse positivity (40×). (b) SMARCB1 immunostain in a choroid plexus carcinoma showing strong nuclear positivity (40×). Retained SMARCB1 staining helps differentiate these tumors from AT/RT, which show loss of SMARCB1 expression. (c) Focal GFAP expression in a choroid plexus carcinoma (20×). (d) Cytokeratin stain (AE1-3) in a choroid plexus carcinoma showing focal immunoreactivity (20×).
Fig. 12.5
Immunohistochemistry for Kir7.1 in choroid plexus tumors. Membranous staining for Kir7.1 in a choroid plexus papilloma (a, 20×) and a choroid plexus carcinoma (b, 20×).
Histological Differential Diagnosis
Depending on the age group, the differential diagnosis of CPTs includes atypical teratoid/rhabdoid tumor (AT/RT), central nervous system (CNS), primitive neuroectodermal tumors (PNET), papillary ependymoma, oligodendroglioma, neurocytoma, papillary tumor of the pineal region (PTPR), and metastases [70–75]. However, the first differential diagnosis to consider is normal choroid plexus. Typically, normal choroid plexus can be distinguished readily enough when the lesion is entirely papillary, with papillae that are not overly cellular and which exhibit a cobblestone surface, and the absence of mitoses (Figs. 12.1a, b). Further, normal choroid plexus epithelial cells express SERCA3, but SERCA3 expression is decreased in CPTs [76].
AT/RT frequently arises in the differential diagnosis, especially in young children [77]. While AT/RT may occasionally demonstrate poorly differentiated epithelial structures, this histologic pattern is generally quite rare. When such cases do arise, the diagnosis may be resolved by immunohistochemical staining for expression of SMARCB1, which is retained in all CPTs (Fig. 12.4b) [73], as well as choroid plexus marker Kir7.1, which stains the majority of CPC but not AT/RT (Fig. 12.5) [78].
Occasionally in pediatric cases, a supratentorial PNET, particularly the medulloepithelioma, may enter into the differential diagnosis. These tumors can usually be distinguished on histologic grounds; they have tubular structures rather than papillary architecture and comprise cells with embryonal rather than epithelial features. Furthermore, medulloepithelioma is characterized by 19q13.42 amplification and LIN28 expression, linking these rare tumors to embryonal tumor with abundant neuropil and true rosettes (ETANTR) [79, 80]. Cribriform neuroepithelial tumor (CRINET) is a rare tumor characterized by cribriform strands and well-defined surfaces, which may be misinterpreted as CPC. Unlike the majority of CPC, however, CRINET is characterized by SMARCB1 loss as well as EMA staining of surfaces [79]. In contrast to AT/RT, prognosis of CRINET seems to be relatively favorable [81].
Papillary ependymomas share the intraventricular location and confusion may arise in CPPs with elongated tumor cells that may give the appearance of overlapping histopathological features (Fig. 12.6a). One important clue to the differential diagnosis is the presence of a delicate basement membrane in CPTs, a feature consistently lacking in ependymomas. While GFAP may be present in both, it is generally stronger and more diffuse in ependymomas. Rarely, synchronous appearance of CPT and ependymoma has been described [82, 83]. PTPR has to be considered in children and young adults with tumors of third ventricular location. The majority of PTPRs can be distinguished from CPTs by absent staining for epithelial membrane antigen and Kir7.1, as well as the presence of distinct MAP-2 immunoreactivity [68].
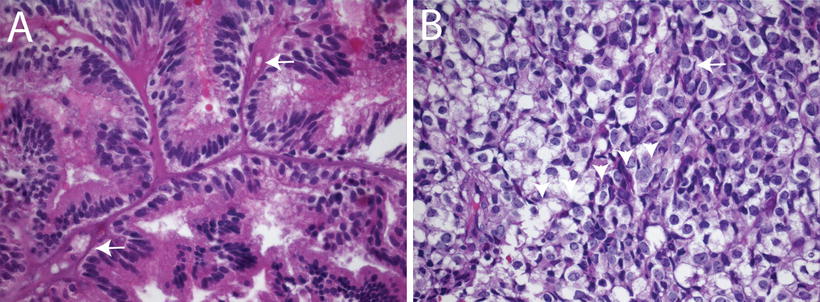
Fig. 12.6
Histologic mimics encountered in choroid plexus tumors. (a) Choroid plexus papilloma showing elongated tumor cells arranged focally around blood vessels reminiscent of an ependymoma (40×). (b) Choroid plexus carcinoma with perinuclear halos surrounding tumor cells mimicking an oligodendroglial neoplasm (40×).
The endolymphatic sac tumor (ELST) is a low-grade carcinoma originating in the ear. These extremely rare tumors are capable of invading the cerebello-pontine angle and might be mistaken for CPTs in this region. Kir7.1 and EAAT-1 (glutamate transporter) are typically positive in CPTs but absent in ELSTs [84]. The choroid plexus is a common site for metastases and this should be considered in any adult with CPTs. Renal cell carcinoma [85], thyroid carcinoma [86, 87], and cholangiocellular carcinoma [88] primaries have all been reported.
Pathogenesis and Molecular Genetics
Pathogenesis
CPTs were the first models for virally induced brain tumors. Simian Virus 40 (SV40), which naturally infects Asian macaques, has been shown to induce CPTs. The virus is capable of transforming human choroid cells in vitro [89–91] and creates CPTs in hamsters and mice in vivo [92–97]. Transgenic mice harboring the SV40 large T-antigen gene develop CPPs by 80–90 days [98, 99]. SV40 is frequently found in human CPTs [100–103]. The T-antigen of the SV40 virus binds to tumor suppressor genes such as p53 [104] and pRB [99]. This suggests virus-induced tumorigenesis. However, an unintended natural experiment that occurred when the vaccine for poliomyelitis was contaminated with the SV40 virus in India did not produce clear evidence of increased incidence of CPTs. It still remains to be conclusively established if SV40 induces CPTs in humans.
Only limited data are available regarding molecular genetic alterations in CPT. Using comparative genomic hybridization, gains of chromosomes 5, 7, and 9 as well as losses of chromosomes 10 and 22q could be demonstrated in CPP. In contrast, CPC mainly showed gains of chromosomes 1, 4, 12, and 20 as well as losses of 5, 18, and 22q [105]. These findings could be extended using high-resolution methods, showing recurrent copy number gains of chromosomes 1, 2, 4, 12, and 20 as well as losses of chromosomes 5, 6, 16, 18, 19, and 22 in CPC. Clustering analysis separated choroid plexus carcinomas into two groups: one characterized by marked losses and the other characterized by gains across the chromosomes. Chromosomal losses of 9, 19p, and 22q were significantly more frequent in younger children (<36 months), whereas gains on chromosomes 7 and 19, and chromosome arms 8q, 14q, and 21q prevailed in older patients [106].
The involvement of the TP53 tumor suppressor gene in CPT patients was first suggested by the occurrence of CPC in families with Li–Fraumeni syndrome [107–109], and by the observation of p53 inactivation in tumor tissues [69]. A Canadian group reported a large CPT population with p53 alterations [110]. A Brazilian study confirmed these finding on a larger scale [111–113] for a specific mutation TP53 mutation: R337H. This TP53 mutation is also linked to adreno-cortical carcinoma. Interestingly, high-resolution single nucleotide polymorphism (SNP) array analysis did reveal extremely high total structural variation in TP53-mutated CPC tumor genomes compared with TP53 wild-type tumors and CPPs [110]. However, in the absence of TP53 germline mutations CPTs may still arise through the same pathway driven by somatic mutations [114]. Even though a close association between TP53 mutation status and nuclear accumulation of p53 protein is often claimed [110], the majority of CPTs show only weak and focal nuclear staining, suggesting that p53 immunohistochemistry might not be a reliable indicator of TP53 mutations in these tumors.
Other molecular events in the pathogenesis of CPT are not yet as well characterized. TP53 mutations are unlikely to be the only event in the pathogenesis of CPTs. Patients with multiple resections show progression of CPTs with a tendency to increasing degrees of malignancy [40], and CPTs may arise from teratomas [115], indicating an accumulation of events leading to the final phenotype. Several other pathways have been suggested to be operative in the biology of CPTs. In mice, over-expression of notch3 initiated the formation of CPTs [116]. Some evidence suggests alterations of notch signaling also occur in human CPT [116, 117].
By comparing gene expression profiles obtained from human CPP cells with that of nonneoplastic choroid plexus epithelial cells, the transcription factor TWIST1 was identified to be highly expressed in CPP and also promoted proliferation and invasion in vitro [118]. Amplification and activating mutations of tyrosine receptor signaling pathways play an important role in the biology of human cancer. In CPC, amplification and over-expression of PDGF receptors has been described [119]. Furthermore, in immortalized choroid plexus epithelial cells, PDGF-BB exhibited a time- and dose-dependent proliferative response, which was significantly attenuated by the tyrosine-kinase inhibitor imatinib [120], providing a rationale for the development of treatments targeting PDGF receptor signaling in CPT.
The role of epigenetic alterations in CPT is also poorly understood. In pediatric brain tumors, human telomerase reverse transcriptase (hTERT) promoter methylation has been shown to be associated with tumor progression and poor prognosis. Methylation of the hTERT promoter has also been reported in the majority of CPCs [121]. The clinical utility of these findings for CPCs remain to be elucidated. Similarly, the prognostic and predictive role of MGMT promoter methylation, which occurs frequently in CPTs [122], remains to be determined.
Clinical Aspects and Treatments
Prognosis and Current Treatment
Due to the low incidence of CPTs, few randomized trials have been conducted [123, 124]. Most data come from individual case reports [50, 125], case series [23, 58, 126–129], or systematic literature reviews [130–133]. These data suggest that histological classification appears to be the most reliable prognostic parameter [134, 135]. Patients with CPPs have long-term survival rates exceeding 95 % when completely resected. In contrast, CPCs in patients treated with surgical resection and radiation therapy have 5-year survival rates of approximately 60 %. Primary and metastatic CPC in infants from Li–Fraumeni families fare even worse, with 5-year survival rates of less than 5 % [130].
Tumor resection is of very high therapeutic value in CPTs [28, 135–141]. In particular, gross total resection was found to be of significant prognostic value in meta-analyses, [130, 134, 141] thereby confirming the institutional experiences of many groups [23, 124, 142]. Meta analyses also confirmed the value of a second resection [143]. However, attempts at radical resection should be made with caution, since the high vascularity of these tumors also translates into a high rate of intratumoral bleeding [136], and other surgical complications such as tension pneumoventricle [144] and hyperacute disseminated intravascular coagulation [145]. Newer surgical techniques might reduce morbidity and mortality. These include endoscopic [137] and combined endoscopic and microsurgical approaches [146]. For tumors of the foramen of Luschka, a telovelar approach has been proposed [147]. Preoperative embolization may reduce the operative risk [6, 50, 148, 149]. In one case the tumor regressed after embolization without the need for resection [150]. Similarly, preoperative intensive chemotherapy may reduce the risk for intraoperative hemorrhage even when the size of the tumor does not shrink significantly [151].
Radiation therapy can increase survival of CPTs in patients old enough to receive therapeutic doses [129, 130, 134, 152, 153]. For example, CPPs may be sensitive to radiation therapy [39] and CPCs may show a survival benefit with craniospinal irradiation [152]. However, long-term sequelae of radiation are particular devastating for the developing brains of young children and limit the use of this modality.
Chemotherapy improved survival rates at least in the subgroup of incompletely resected CPC [42, 132]. The five most frequently used drugs are cisplatin, vincristine, cyclophosphamide, carboplatin, and etoposide. Of those, etoposide (VP16) was most frequently used in protocols and had the most convincing survival benefit in various multivariate analyses [133]. More recently reports suggest temozolomide is less promising [125]. In a prospective clinical trial, CPT-SIOP-2000, cyclophosphamide was found equally effective to carboplatin. As a result of these experiences, the benefit of chemotherapy (including high-dose chemotherapy recently reported for an adult patient [154] is becoming more widely accepted, at least for young children [135, 153, 155, 156]. However, intensive chemotherapy is associated with its own risks, and fatal complications have been described [136]. These studies highlight the need for better targeted and less toxic agents.
The influence of germline TP53 mutations on prognosis and efficacy of treatment remains controversial. A large series of patients treated mainly with intensive chemotherapy including ifosfamide etoposide carboplatin (ICE) show a significantly worse prognosis in patients with Li–Fraumeni syndrome [110]. A second report of patients treated with various chemotherapeutic protocols, among them head start III, described long-term survivors among the Li–Fraumeni population [157]. However, data from the Brazilian family with TP53-R337H mutations failed to show statistically significant differences in survival on treatment [111]. Finally, in the international CPT study, there was no significant difference between Li–Fraumeni and non-Li–Fraumeni families. These differences in results prompt further prospective evaluations.
Unique biological features of CPTs may provide leads to novel therapeutic approaches. The blood–brain barrier is located typically in the vascular wall and is characterized by tight junctions between endothelial cells. In contrast, choroid plexus capillaries are leaky. As this feature of leaky blood vessels is maintained in CPTs, systemic medication may reach tumor cells even among the most differentiated CPTs without hindrance from the blood–brain barrier. The normal choroid plexus also functions as an immunological gate to the CNS, including interferon-γ signal mediated entry of circulating leucocytes for immune surveillance [158], and IL-6 production [159]. It is possible that these immune pathways could be leveraged to develop novel therapeutic approaches against CPTs in the future.
Summary
CPTs are tumors arising from the choroid plexus and based on histologic criteria are classified as CPP, APP, and CPC, which correspond to WHO grades I, II, and III, respectively. CPTs occur in all ages but are more common in childhood, peaking in incidence during the first decade of life. Histological grading remains a key prognostic factor and several ancillary immunohistochemical tests can aid in establishing their diagnoses. CPPs are usually treated with surgical resection, whereas a combination of surgical resection and/or chemo/radiation therapy may be used for higher-grade tumors. The biology of CPTs is poorly understood. Factors implicated in the pathogenesis of CPTs include alterations in p53- and SV40-induced viral transformation. However, the molecular genetics of CPT initiation and progression have not been otherwise elucidated and should provide fruitful avenues for future research.
References
1.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109.PubMedCentralPubMed
2.
Jeibmann A, Hasselblatt M, Gerss J, Wrede B, Egensperger R, Beschorner R, Hans VH, Rickert CH, Wolff JE, Paulus W. Prognostic implications of atypical histologic features in choroid plexus papilloma. J Neuropathol Exp Neurol. 2006;65:1069–73.PubMed
3.
Zulch, ed. Brain tumors: their biology and pathology. New York: Springer; 1957.
4.
Stagno V, Mugamba J, Ssenyonga P, Kaaya BN, Warf BC. Presentation, pathology, and treatment outcome of brain tumors in 172 consecutive children at CURE Children’s hospital of Uganda. The predominance of the visible diagnosis and the uncertainties of epidemiology in sub-Saharan Africa. Childs Nerv Syst. 2014;30:137–46.PubMed
5.
Wilhelm M, Hirsch W, Merkenschlager A, Stepan H, Geyer C, Kiess W. A rare case of congenital choroid plexus carcinoma. Pediatr Hematol Oncol. 2012;29:643–6.PubMed
6.
Ditz C, Nowak G, Koch C, Merz H, Tronnier V. Atypical choroid plexus papilloma in a newborn: prenatal diagnosis, preoperative tumor embolization, and resection. J Neurol Surg A Cent Eur Neurosurg. 2013;74:59–63.PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


