Chapter 11
Chemotherapy of infectious diseases
11.1 Introduction
In the treatment of infections, the goal is to kill the microbes, or at least to disrupt their ability to propagate, so that the immune system can get the better of them. While this is simple in principle, the difficulties in practice are twofold. Firstly, the drugs must kill the microbes but spare the host; this is the principle of selective toxicity. Secondly, microbes tend to develop resistance to drugs that are initially effective. The problem of resistance is both grave and unavoidable, since microbes are subject to genetic variation, and each drug use, medically justified or not, will select for more resistant variants. Therefore, antimicrobial drug discovery may have to continue in perpetuity.
11.2 Diversity of infectious pathogens
- Bacteria
- Fungi
- Parasites—eukaryotes other than fungi
– Protozoa—unicellular
– Metazoa—multicellular
- Viruses
Notes: Among the parasites, the category “protozoa” is a bit of a historical relic. When infectious agents were first identified, the genetic and biochemical relationships between them were not understood, and newly discovered pathogens were classified somewhat haphazardly and mostly according to morphological criteria. The term “protozoa” means “primitive animals”. However, as shown in the next slide, the protozoa are rather distantly related to humans and other real animals, and even to one another.
The large evolutionary distance between protozoa and humans corresponds to significant differences in cellular biochemistry, some of which can be exploited for chemotherapy.
11.2.1 The tree of life, slightly pruned
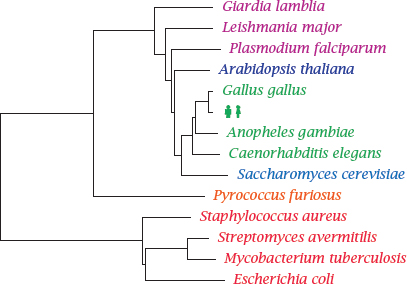
Notes: This phylogenetic tree shows Homo sapiens in the company of some pathogens and reference organisms. Arabidopsis is a plant, and Saccharomyces is a yeast; both are more closely related to humans than are Plasmodium, Leishmanial and other single-celled eukaryotic parasites that have been lumped in with the protozoa. The nematode Caenorhabditis elegans is not a pathogen itself but is included as a proxy for human worm pathogens such as Wuchereria bancrofti or Ascaris lumbricoides.
Pyrococcus furiosus1 belongs to the Archaebacteria. These organisms have a prokaryotic cell structure but are phylogenetically closer to us than to the Eubacteria, to which all pathogenic bacteria belong. Among these, Staphylococcus aureus represents the Gram-positives, whereas Escherichia coli represents the Gram-negatives. These two major groups have different cell wall structures (see slide 11.4.1), which causes differences in antibiotic susceptibility. Mycobacteria such as Mycobacterium tuberculosis, which are genetically almost equidistant from both, have another distinct cell wall architecture that is particularly hard to penetrate for antibiotics.
The cell wall structure of the streptomycetes resembles that of the Gram-positives, but they are genetically closer to the mycobacteria. They occasionally occur as pathogens but are most notable as purveyors of antibiotics; the majority of all known natural antibiotics is produced by member species of Streptomyces or related genera.
11.2.2 Drug targets for antimicrobial therapy
- Macromolecules that occur only in the pathogen but not the human host. Examples:
– the bacterial cell wall (penicillin)
– de novo synthesis of folic acid (sulfonamides)
- Macromolecules that occur in both humans and the pathogen but are structurally divergent. Examples:
– ribosomes (chloramphenicol)
– dihydrofolate reductase (trimethoprim)
– DNA topoisomerase (ciprofloxacin)
Notes: This list of examples shows that both pathogen-specific drug targets and those that are shared but divergent are important and viable in clinical practice.
In recent years, the genomes of many pathogens have been sequenced and searched for genes that are essential for the life of the pathogens, and at the same time do not have counterparts in the human genome. While the products of such genes should indeed make good candidate drug targets, limiting the search for targets to such pathogen-specific macromolecules seems unduly narrow, given the proven value of many drugs that act on shared but divergent targets.
Both shared and pathogen-specific drug targets tend to be more abundant in pathogens with greater evolutionary distance from humans. Thus, the number of suitable targets in bacteria is greater than that in the other types of pathogens.
11.2.3 Structures of folic acid and of three inhibitors of dihydrofolate reductase
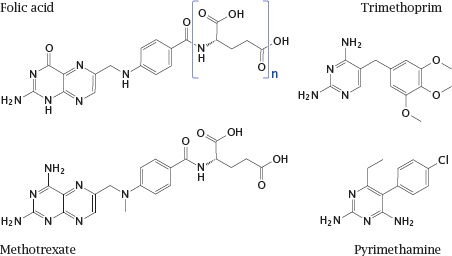
Notes: Dihydrofolate reductase is an excellent example of a shared drug target. It is essential in all organisms for sustaining the folic acid-mediated transfer of singlecarbon groups in biosynthetic reactions. Nevertheless, the enzyme molecules found in various organisms are sufficiently different to allow their selective inhibition with various chemotherapeutic drugs.
Trimethoprim inhibits the bacterial enzyme. It acts synergistically with sulfonamides, which inhibit the bacterial de novo synthesis of folic acid (see slide 1.3.3). Trimethoprim is typically used in combination with a sulfonamide.
Pyrimethamine inhibits the enzymes of protozoans such as Toxoplasma gondii and Plasmodium falciparum. The human enzyme is not significantly inhibited by either but is inhibited by methotrexate, which is used in anticancer and immunosuppressive therapy.
11.2.4 Microbial resistance mechanisms
- Mechanisms affecting the target:
– Structural alteration / mutation
– Compensatory overexpression
- Mechanisms affecting the drug:
– Reduced uptake
– Active extrusion
– Enzymatic inactivation
Notes: Resistance to chemotherapy occurs with all kinds of pathogenic microbes. While it poses obvious and significant practical problems, it is also a fascinating microcosm of Darwinian evolution through mutation and selection. Larger organisms evolve too slowly for us to observe; with drug resistance in microbes, we get a time-lapse view of evolutionary adaption. This is due to both the short generation time of microbes and the rigorous nature of the selection.
This slide lists the principal mechanisms of resistance that apply to all classes of pathogenic microbes. We will see specific examples for most of these mechanisms in the following sections.
11.3 Overview of antibacterial chemotherapy
- Targets
– Cell wall
– Ribosomes
– Enzymes related to cell division
– Intermediate metabolism
- Antibiotic resistance
– Bacteria have short generation times—fast de novo evolution of resistance
– Resistance genes exist in producers of antibiotics—can spread to pathogenic bacteria by gene transfer
Most antibacterial drugs are antibiotics, that is, natural compounds isolated from other microbes, or derivatives thereof. While penicillin was famously isolated from a mold, most antibiotics—for example, tetracyclins, aminoglycosides like streptomycin, and macrolides like erythromycin—are actually produced by Streptomyces species or related soil bacteria. Since these producer bacteria must be resistant to their own poisons, it follows that mechanisms and genes for bacterial resistance must exist for any of these natural antibiotics. Such genes may migrate to clinical pathogens and spread among them if we apply the proper selection pressure through the medical use and misuse of those antibiotics.
Some antibacterial compounds are indeed fully synthetic; we have already seen sulfonamides and trimethoprim as examples. With these agents, resistance genes may not exist a priori; however, resistance often emerges through spontaneous mutations that sharpen the target enzymes’ ability to discriminate between the drugs and the proper substrates.
With both natural and synthetic antibiotics, an important strategy to prevent, or at least delay, the emergence of resistance is combination therapy. When several drugs are combined that each alone are able to kill the pathogen, and each of which addresses a different target, the pathogen would have to simultaneously modify all targets in order to survive. With increasing number of agents simultaneously applied, this rapidly becomes unlikely.2
11.3.1 Gene transfer mechanisms in bacteria
- Transformation: cellular uptake of naked DNA
- Conjugation: plasmid-encoded active transfer between bacterial cells
- Transduction: gene transfer mediated by bacteriophages
- Transposons: transfer of genes between carrier DNA molecules (chromosomes, plasmids)
Notes: Transformation, conjugation and transduction all work within and also between bacterial species.3 Transfer between species is important in the migration of resistance genes from producer strains, or other soil bacteria that are naturally exposed and have developed resistance to a given antibiotic, to pathogenic bacteria.
Resistance genes are often carried by transposons, which are mobile genetic elements that can “hop” between different carrier molecules. Given the right selection conditions, multiple resistance transposons may wind up on a single plasmid molecule. Such a plasmid will then cause resistance to several unrelated antibiotics to spread all at once, even if only one of them is actually used in a clinical setting. The first such multiresistance plasmids were observed only about ten years after the start of the antibiotic era.
11.4 Antibiotics and the bacterial cell wall
In contrast to human cells, which are simply delimited by their cytoplasmic membranes, most bacteria have cell walls that consist of one ore more protective layers stacked on top of their cytoplasmic membranes. While these cell walls may protect the bacteria from antibiotics, they also provide targets for chemotherapy. Note that some bacteria—mycoplasmas, and the vegetative forms of rickettsias and chlamydias—have no cell wall at all and therefore are not susceptible to the agents discussed in this section.
11.4.1 Bacterial cell wall structure
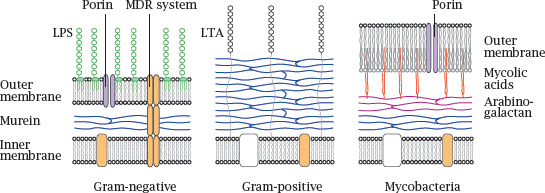
Notes: The innermost layer of a bacterial cell wall consists of murein or peptidoglycan, a meshwork of polysaccharide strands crosslinked by oligopeptides. Gram-negative bacteria have a comparatively thin peptidoglycan layer (blue) that is surrounded and protected by an outer membrane. The outer leaflet of this lipid membrane consists mostly of lipopolysaccharide (LPS, green), which is also known as endotoxin.4 Porins in the outer membrane facilitate diffusion of small polar solutes. Multidrug resistance (MDR) proteins that extrude antibiotics from the cell may be located in the cytoplasmic membrane alone or span both membranes.5
Gram-positive bacteria lack an outer membrane but have a much thicker murein layer, which is decorated with lipoteichoic acids (LTA). The lack of an outer membrane makes them more amenable to penicillin (see slide 1.3.10) and many other antibiotics.6
In mycobacteria, the murein layer is surrounded by arabinogalactan polysaccharide, to which branched, long-chain fatty acids (mycolic acids, red) are attached. The mycolic acids act as anchors for a particularly thick, wax-like and impenetrable outer membrane.
Because of their sturdy cell wall,7 mycobacteria have always been among the most difficult microbes to treat—although decades of selection have bred some real champions of resistance among the Gram-positives and Gram-negatives also.
11.4.2 Action mechanism of isoniazid (INH)
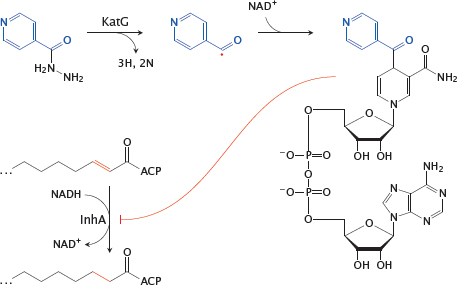
Notes: Isoniazid is effective against the pathogen Mycobacterium tuberculosis. It inhibits the synthesis of mycolic acids, the long-chain fatty acids that are characteristic and essential components of the mycobacterial cell wall. This inhibition occurs in a rather unique manner.
Inside the mycobacterial cell, INH is activated by the oxidative enzyme KatG8 to a radical form. This radical then reacts with NAD+. The adduct inhibits InhA, an enoyl-CoA reductase involved in mycolic acid synthesis.
11.4.3 Outline of bacterial murein synthesis
Notes: Murein, or peptidoglycan, is found in the cell walls of Gram-positives, Gram-negatives, and mycobacteria. Inhibitors of various enzymes in the murein synthesis pathway are active against member species of all three classes.
The synthetic pathway involves the following stages: Phosphoenolpyruvate (PEP) supplies a lactate residue (Lac) that is attached to N-acetylglucosamine, which yields N-acetyl-muramic acid (1). Onto the latter, a pentapeptide is built in a series of ATP-activated reactions. The free end of this peptide contains two D-alanine residues that are supplied by alanine racemase (2) and D-alanine ligase (3). This nascent building block is transferred to the lipid carrier undecaprenol phosphate (4) and subsequently extended by another molecule of N-acetylglucosamine and five glycine residues.
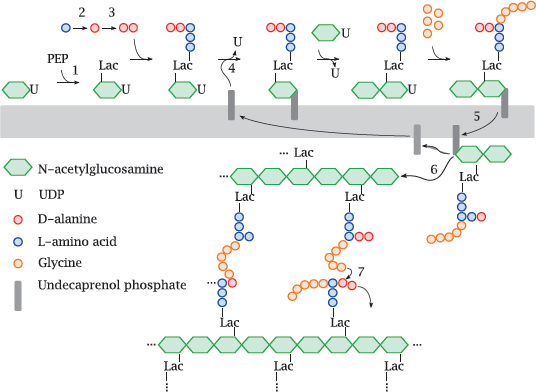
The transglycosylase and transpeptidase activities are both located on the same enzyme protein, variously referred to as muramyl-transpeptidase or penicillin-binding protein.
11.4.4 Fosfomycin resembles both phosphoenolpyruvate and glycerophosphate
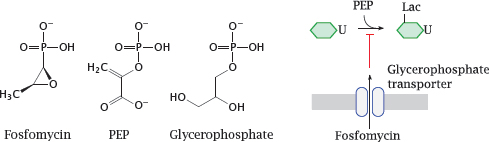
Notes: Fosfomycin is an antimetabolite of phosphoenolpyruvate (PEP) in the first step of murein precursor synthesis. Considering its structure, it may not surprise you to hear that the reaction with the target enzyme is covalent and involves the thiol group of a cysteine residue [73].
For uptake across the cytoplasmic membrane, fosfomycin piggybacks on a transport protein that mediates uptake of glycerophosphate. This transporter is not essential for the bacterial cell; therefore, mutations that inactivate the transporter tend to cause rapid development of resistance under therapy. Fosfomycin can therefore only be used in combination with other antibiotics that are less prone to rapid evolution of resistance.
11.4.5 Cycloserine inhibits alanine racemase and D-alanine ligase
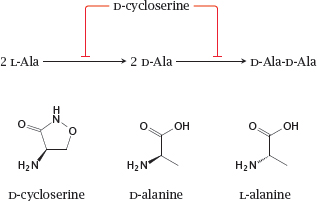
Notes: D-cycloserine is an antimetabolite of D-alanine in both the alanine racemase and the D-alanine ligase reactions. It is used mostly against mycobacterial infections, though active in principle against other types of bacteria also.
Interestingly, D-cycloserine is also a partial agonist at the sole glycine-specific subunit of the NMDA-type glutamate receptor (see section 6.11). As such, it has been tried therapeutically in various neurological and psychiatric conditions [74].
11.4.6 β-Lactam antibiotics resemble the substrate of the transpeptidase reaction
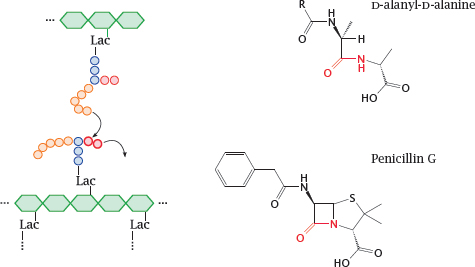
Notes: We now skip ahead to the last step of murein synthesis, that is, the transpeptidase reaction that crosslinks the assembled linear strands of murein and thereby confers stability to the cell wall.9
11.4.7 Reactions of penicillin G with transpeptidase and β-lactamase
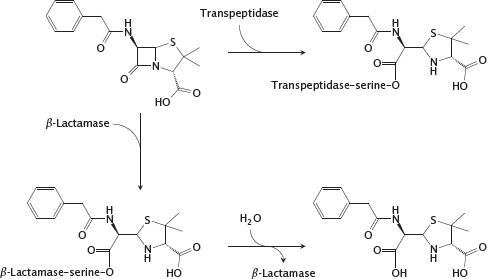
Notes: The shared structural feature that gave the class its name is also at the heart of antibacterial action. The sterically strained four-membered β-lactam ring is spring-loaded like a mousetrap and readily reacts with the catalytic serine residue of the transpeptidase enzyme. The enzyme is unable to free itself from the covalent modification and remains irreversibly inactivated.
The most prevalent mechanism of bacterial resistance to β-lactams is enzymatic inactivation by β-lactamases. The enzymes in one of the two major classes of β-lactamases also contain a catalytic serine. In contrast to the transpeptidase, such a β-lactamase regenerates its free serine through hydrolysis and thus can successively degrade a large number of antibiotic molecules. Some of these enzymes can be inhibited with β-lactamase inhibitors. The β-lactamases in the second class are metalloenzymes. In contrast to the serine enzymes, they do not form covalent intermediates, and no clinically useful inhibitors are currently available.
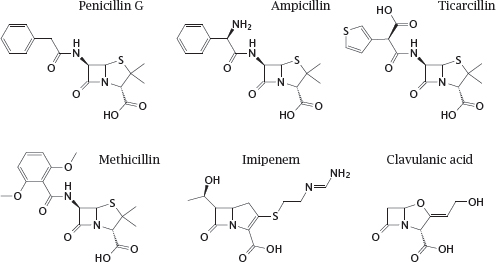
11.4.8 Structures of β-lactam antibiotics (1)
Notes: The first β-lactam to be used clinically, penicillin G, is active almost exclusively against Gram-positive bacteria, since it fails to penetrate the outer membrane of most Gram-negatives. Other β-lactams that were developed later have progressively extended the spectrum to include most Gram-negatives and even some mycobacteria. Some of these derivatives are also, to varying degrees, protected from inactivation by bacterial β-lactamases.
Penicillin G itself is readily cleaved by β-lactamases, and while it was active on almost all strains of Staphylococcus aureus when first introduced, more than 90% of all clinical isolates of this pathogen now possess β-lactamases that render them resistant. To counter this resistance mechanism, semisynthetic drugs such as methicillin were developed, which escape cleavage by the enzyme through steric obstruction. This obstruction also renders such drugs about ten times less active against the muramyl-transpeptidase target; but because of the generally very high therapeutic index of the penicillins, this does not hamper their clinical utility. However, methicillin-resistant S. aureus strains (MRSA) have emerged and become increasingly widespread. Through horizontal gene transfer from another staphylococcal species, these strains have acquired a peculiar variant of the transpeptidase that does no longer binds methicillin and related compounds. MRSA are also resistant to most other β-lactams; they are, however, susceptible to ceftobiprole (see next slide).