Chemical equilibrium
As stated by the IUPAC, equilibrium is the state of a system in which the macroscopic properties of each phase of the system become uniform and independent of time. If the temperature is uniform throughout the system, a state of thermal equilibrium has been reached; if the pressure is uniform, a state of hydrostatic equilibrium has been reached; and if the chemical potential of each component is uniform, a state of chemical equilibrium has been reached. If all of these quantities become uniform the system is said to be in a state of complete thermodynamic equilibrium.
11.1 Chemical potential and Gibbs energy of a reaction mixture
The chemical potential of a pure substance is defined as

In words, the chemical potential shows how the Gibbs energy of a system changes as a substance is added to it. For a pure substance, the Gibbs energy is simply G = nGm, therefore,

The chemical potential of a pure substance is the same as the molar Gibbs energy.
From Eq. (9.38) we write the chemical potential of an ideal gas


For a system composed of several chemical species in the thermodynamic limit

Remember what we mean by the thermodynamic limit: for a sample of material that is of such large dimensions that its size and shape no longer affect its properties. For an ideal gas, this simply means that we have a large volume. To write Eq. (11.5) more generally we need to explicitly write a term that includes a contribution from the interfaces present in our system. Consider for example a system composed of a solid A in contact with its vapor B. The Gibbs energy of the system is

where γAB is the Gibbs energy per unit area of interface and A is the area of the interface. When the number of surface atoms is small, this last term can be neglected. As we have seen in Chapter 5, the ratio of the number of surface atoms to total atoms is a function of the size of the sample, and this ratio only exceeds a non-negligible number, say 10−3, at radii of ≥100 nm for spherical particles. Therefore, as long as our system is larger than ∼100 nm, we can neglect the interfacial terms. However, if we are dealing with colloids or many systems of interest in soft matter physics (e.g., micelles, bilayers, etc.), the Gibbs energy per unit surface area is significant and must be considered.
A system evolves until its Gibbs energy has obtained one constant value that is a minimum. By definition then, a system also seeks to minimize its chemical potential. For a given species, transport will occur spontaneously from a region of high chemical potential to one of low chemical potential. The flow of material will continue until the chemical potential has the same value in all regions of the mixture. This property of the chemical potential defines chemical equilibrium.
At equilibrium, the chemical potential of each individual species is the same throughout a mixture.
11.2 The Gibbs energy and equilibrium composition
11.2.1 Gibbs energy and chemical potential determine the direction of change
Consider the equilibrium between two species A and B.

The reaction Gibbs energy can be expressed as the difference between the chemical potential of the products and the reactants

Specifically for the simple reaction in this example,

The Gibbs energy change is the control parameter that tells us how the system will evolve. The spontaneous direction of change is the direction that requires no work to be done for the system to proceed in that direction.

The system will evolve from high chemical potential to low chemical potential. When the chemical potential of the reactants and products is the same, equilibrium has been achieved. We have to use drG and not ΔrG as the criterion for spontaneous direction of change to be absolutely correct. This is illustrated in Fig. 11.1 Even though point C lies at lower Gibbs energy than point A – that is, ΔrG(A→C) < 0 – the system will not evolve from A to C. This is because there is a minimum at B along the way. The system evolves from A to B. Along this path drG < 0 everywhere until B is reached. At B, drG = 0. After B, drG > 0 and the system will not continue to proceed uphill. Note the pathological point D, where the Gibbs energy is equal to that at C. At this point ΔrG(D→C) = 0, but clearly drG < 0 and the system is not at equilibrium.

Figure 11.1 The sign of the slope of the Gibbs energy versus extent of reaction determines the direction of spontaneous change. The system will evolve as long as there are no kinetic barriers to the minimum Gibbs energy for the system. Regardless of whether the system starts at point A or point C, the direction of spontaneous change is towards point B.
Note what spontaneous means in a thermodynamic sense. Spontaneous means that the process occurs irreversibly and it will not go back unless we do work on the system. drG > 0 means the process is impossible without application of work; that is, the reverse happens. drG ≠ 0 means the extent of reaction has to change, dξ ≠ 0. drG = 0 does not mean that nothing happens; it means that the extent of reaction does not change, dξ = 0, because the rates of the forward and reverse processes are equal. Thus, spontaneous in the thermodynamic sense has a more nuanced meaning than is encountered beyond thermodynamics.
11.2.2 Ideal gas equilibria
Take A and B to be ideal gases. Substitute from Eq. (9.38) for the chemical potential to obtain,

The standard reaction Gibbs energy is defined as
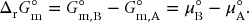
The reaction quotient Q is the ratio of partial pressures of products, each raised to the power of its stoichiometric coefficient, divided by the partial pressures of the reactants, each raised to its stoichiometric coefficient. Each partial pressure must also be divided by the standard state pressure to ensure that the reaction quotient is dimensionless. For the reaction of A to B,

This us allows us to write Eq. (11.9) more generally as
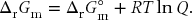
Previously, we have shown that ΔrG°m can be calculated from standard Gibbs energies of formation according to

At equilibrium ΔrGm = 0, and the reaction quotient is equal to the equilibrium constant K. Therefore we can write

This is one of the most important equations in thermodynamics as it links thermochemical data to the value of the equilibrium constant. It is known as the reaction isotherm.
It is essential here that the difference between ΔG and ΔG° is understood. The difference is defined by the difference between the reaction quotient Q and the equilibrium constant K. Q is tied to ΔG. K is linked to ΔG°. Q and ΔG refer to arbitrary composition. The equilibrium constant K refers specifically to the composition at equilibrium, and ΔG° refers specifically to each species being in its standard state. Large values of the equilibrium constant, K ≫ 1, are associated with negative values of ΔG°. This favors the products at equilibrium. Small values of K, K ≪ 1, are associated with positive values of ΔG°. Reactants are favored at equilibrium. In contrast, the meaning of ΔG is to tell us the direction the system evolves spontaneously from the current composition defined by Q.
To emphasize the difference between ΔG and ΔG°, the affinity of reaction A, defined by

has been introduced. Thus, when A is positive, the reaction proceeds in the forward direction, and when A is negative it proceeds in the reverse direction. Unfortunately, A is also the preferred symbol for the Helmholtz energy.
Equation (11.14) is not limited to ideal gases. Indeed, we can write a generalized reaction quotient in terms of fugacities (real gases) or activities (real mixtures) and derive the same equation. The relation between K and ΔfG°m is general and holds for all real and ideal systems.
11.2.2.1 Directed practice
Calculate K for the ammonia synthesis reaction at 298 K, assuming all gases to be ideal and ΔfG°m for NH3 to be −16.4 kJ mol−1.
[Answer: K = 6.09 × 105]
11.2.2.2 Directed practice
Determine K at 298 K for the reaction C2H4(g) + H2(g)  C2H6(g), given that ΔfG° for C2H4(g) and C2H6(g) are 68.4 and −32.0 kJ mol−1, respectively.
C2H6(g), given that ΔfG° for C2H4(g) and C2H6(g) are 68.4 and −32.0 kJ mol−1, respectively.
[Answer: K = 3.96 × 1017]
11.2.2.3 Directed practice
A mixture of CO(g), H2(g) and CH3OH(g) at 500 K with p(CO) = 10 bar, p(H2) = 1 bar and p(CH3OH) = 0.1 bar is passed over a catalyst. Can more methanol be formed? ΔrG°m = −21.21 kJ mol−1.
11.3 The response of equilibria to change
A chemical equilibrium is a system of reactants and products that are coupled to each other by reversible reactions. Any change to the system variables, such as p, T, V and composition, will cause the system to react in response to this change. This idea is stated as Le Châtelier’s principle.
A system at equilibrium, when subjected to a disturbance, responds in a way that tends to minimize the effect of the disturbance.
Consider methanol synthesis from syn gas (syn gas is short for synthesis gas, a mixture of CO and H2 that can be made from gasification of coal, biomass or other hydrocarbons),

Suppose the reaction is run at 298 K but at low enough pressure so that all species are in the gas phase. By inspection of the stoichiometry, we see that 3 mol of gas are converted to 1 mol of gas. This corresponds to an unfavorable entropy factor. Indeed, upon calculation we find ΔrS°m = −219 J K−1 mol−1. Applying Le Châtelier’s principle, increasing the pressure on the system will lead it to relieve this stress by proceeding in the forward direction. Decreasing the amount of substance of gas (at constant T and V) lowers the pressure. If the pressure is increased enough, the methanol will eventually condense, lowering the pressure even further. By inspection, one cannot tell easily what the enthalpy change is. Calculation reveals ΔrH°m = −90.6 kJ mol−1. For an exothermic reaction such as this, heat can be thought of as a product. Thus, raising the temperature (at constant p and V) ought to push the system to the left towards reactants. The system reacts to ‘use up the heat’ added to the system by raising T. Conversely, raising the temperature of an endothermic reaction tends to push it towards the products.
Warm and fuzzy statements like Le Châtelier’s principle are helpful to organize our thoughts and spark intuition, but this is physical chemistry and we want to understand the basis of these principles. We can do this by understanding the dependence of the Gibbs energy and the equilibrium constant on changes in variables such as T and p.
11.3.1 Response to temperature
The control parameter that we monitor to tell us how a system will evolve is the Gibbs energy. At constant temperature and pressure a system evolves to minimize the Gibbs energy. The spontaneous direction of change is always the path that leads to a lowering of Gibbs energy. As derived above, the Gibbs energy is related to composition from the equilibrium constant through

Now, substitute from the fundamental relationship of Gibbs energy to enthalpy and entropy

and solve for ln K to obtain

This is known as the integrated form of the van’t Hoff equation, also referred to as the van’t Hoff isochore. R is the gas constant and T the absolute temperature. We see that the Gibbs energy of reaction and the equilibrium constant are inherently temperature-dependent.
Of all the equations in thermodynamics, the two you should memorize and know immediately off the top of your head are Eqs (11.16) and (11.17). They are important in their own right and because so much can be derived from them. In this case, we have used them to derive the van’t Hoff equation, which is of particular importance because it relates K – a quantity that is directly related to concentrations and the reaction stoichiometry – to the thermodynamic quantities ΔrH°m and ΔrS°m. These quantities are measured directly by thermochemical methods. For some systems, thermochemical methods may be easily performed over a certain temperature range but not over others. For other systems, concentrations might prove much easier to measure than heat flows. Equation (11.18) allows us to pick the experiments that are most accurate and reliable. It also allows for comparisons between methods to enhance accuracy and test assumptions.
Looking at the van’t Hoff equation in more depth, we see that measurement of K as a function of temperature can lead to values for ΔrH°m and ΔrS°m. As is our want, we can turn this equation into a straight line if we plot ln K versus 1/T. If ΔrH°m is constant, this so-called van’t Hoff plot will be linear with slope = −ΔrH°m/R and intercept ΔrS°m/R. An example of a van’t Hoff plot is shown in Fig. 11.2.

Figure 11.2 The self-ionization of water from 0 °C to 100 °C. Slight curvature in the plot of ln Kw versus 1/T indicates that ΔrH°m and ΔrS°m are slightly temperature-dependent over this temperature range. The temperature dependence of Kw means that the pH of neutral water is also T-dependent, as discussed further in Section 11.5.1.
First, a word about slopes and intercepts in general. In many experiments absolute calibration is difficult but relative calibrations are easy. While signals are linear in a certain parameter, they are often subject to an offset (the ‘zero’ in the machine is not precisely known). The determination of a slope is unaffected by an offset as long as it is constant. In other words, as long as we are adding a constant amount to all points on a line (or any curve for that matter), it is immaterial if the line is a bit higher or a bit lower on our graph, the slope is still the same. On the other hand, the presence of an offset directly changes the value of the intercept. Experimentally, slopes are more reliably determined than intercepts not only because of this insensitivity to offsets, but also because evaluation of the slope does not require an extrapolation. Historically, therefore, preference has been given to methods and equations that rely on slopes rather than intercepts. With increasing experimental precision and accuracy – for instance, knowledge of absolute concentrations rather than merely signals that are proportional to concentration – both slopes and intercepts become more reliably accessible.
Is the integrated form of the van’t Hoff equation given in Eq. (11.18) good enough? The answer for that depends on the quality of the experiments that are performed (how big the experimental uncertainties are) and what we are attempting to learn (how accurately we can interpret the data). We know that in general ΔrH°m and ΔrS°m are temperature-dependent. Strictly speaking, a van’t Hoff plot should never be linear. Nonetheless, most van’t Hoff plots are evaluated with this simple linear form; furthermore, most look linear. Over a small enough range of x values, any curve is linear. Reactions performed in liquid solvents can only be performed over a range in which the solvent is stable. For water, this range is no more than ΔT ≈ 100 K, while even a more stable solvent such as toluene can only be used over a range of ΔT ≈ 200 K. Practically speaking, the ranges will be much smaller since solvent evaporation over the course of the experiment must be avoided. As shown in Fig. 9.5, temperature ranges of a few tens of kelvin are simply insufficient to observe a significant change in ΔrH°m. If, on the other hand, a reaction can be followed in the gas phase (or perhaps in a liquid metal or molten salt) over hundreds of kelvin, then the temperature dependence will be significant.
A significant change is one that has a measurable effect on the outcome of experimental results beyond experimental uncertainty. If your experiments represent the first characterization of a new reaction involving novel compounds, then you are probably happy that you can measure the equilibrium constant over any temperature range at all. Your accuracy may only be 5–10 kJ mol−1. Linear analysis of a van’t Hoff plot is completely justified. In contrast, if generations of experiments have been performed with progressively better signal-to-noise ratios and lower experimental uncertainty, and you are aiming to test the limits of our understanding of the intricate nature of the system on a microscopic level, then your analysis needs to be as accurate as possible.
The effects of temperature are more generally expressed in terms of the differential form of the van’t Hoff equation, which is written in two ways

Upon separation and integration we obtain,

We now substitute the functional form of ΔrH°m that we need for our level of theoretical understanding. If we take the enthalpy change to be independent of temperature, we obtain a result that is equivalent to Eq. (11.18),

If sufficient experimental information is available, we can substitute an appropriate polynomial form for the temperature dependence of ΔrH°m, such as that shown in Eq. (9.47). On the other hand, the temperature dependence of ΔrG°m, and therefore K, are probably much more easily approximated by using Eq. (11.17), and substituting in directly the temperature-dependent values of the enthalpy and entropy changes, as is done in the following example. Nevertheless, the integrated form of the van’t Hoff equation in the form of Eq. (11.21) is quite useful because it allows us to use measurements of K as a function of T to determine ΔrH°m. Equation (11.21) predicts that a plot of ln K versus 1/T yields a straight line with slope − ΔrH°m/R.
11.3.1.1 Example
Using data from the CRC Handbook of Chemistry and Physics, plot ΔrG°m versus T over the temperature range 0 to 100 °C for the self-ionization of water. Compare the accuracy of the Gibbs–Helmholtz expression in Eq. (9.37) to a calculation from Eq. (11.17) that includes the temperature dependence of ΔrH°m from Eq. (9.50) and ΔrS°m from Eq. (9.66).
The self-ionization of water can be represented by the reaction

The CRC Handbook of Chemistry and Physics reports values of ln Kw over this temperature range that are plotted in the form of a van’t Hoff plot in Fig. 11.2. The self-ionization constant of water is related to the Gibbs energy of reaction, as is any equilibrium constant, by
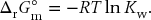
The CRC Handbook of Chemistry and Physics also reports the values in Table 11.1, which are used to calculate ΔrG°m, ΔrH°m, and ΔrS°m for the reaction at T1 = 298 K. These are calculated from  and similarly for ΔrH°m, and ΔrS°m.
and similarly for ΔrH°m, and ΔrS°m.
Table 11.1 Thermodynamic parameters related to the self-ionization of water. All values at 298.15 K taken from the 96th edition of the CRC Handbook of Chemistry and Physics.
| Species | ΔfG°m/kJ mol−1 | ΔfH°m/kJ mol−1 | ΔfS°m/J K−1 mol−1 | Cp,m°/J K−1 mol−1 |
|---|---|---|---|---|
| H2O(l) | −237.1 | −285.830 | 69.95 | 75.3 |
| H+(aq) | 0. | 0. | 0. | 0. |
| OH−(aq) | −157.2 | −230.015 | −10.90 | −148.5 |
First, we generate the blue curve in Fig. 11.3 using Eq. (9.36) with ΔrG°m(T1) = 79.9 kJ mol−1 and ΔrH°m(T1) = 55.815 kJ mol−1 calculated from the appropriate formation values in Table 11.1, and take T1 = 298.15 K. Note that the Gibbs–Helmholtz equation is mathematically equivalent to assuming that ΔrH°m and ΔrS°m are constant and using ΔrG°m = ΔrH°m − T ΔrS°m to calculate ΔrG°m(T2). The blue line is exact at 298 K, but increasingly deviates from the true value (the red square points) as temperature increases. Because the data is of such high quality, clear curvature is recognizable in the plot, which shows that the deviation from the Gibbs–Helmholtz equation is symptomatic of a failure in the assumption that ΔrH°m is constant.

Figure 11.3 The Gibbs energy change for the self-ionization of water exhibits noticeable curvature from the temperature dependence of the enthalpy and entropy of reaction. The blue line is the result of the Gibbs–Helmholtz equation assuming that the enthalpy change is constant at its values from 298 K. Values are taken from the 96th edition of the CRC Handbook of Chemistry and Physics.
The red curve in Fig. 11.3 is obtained by calculating ΔrG°m from ΔrG°m = ΔrH°m − T ΔrS°m substituting ΔrH°m(T2) = ΔrH°m(298 K) + (T2 − 298.15) ΔrC°p, m for the enthalpy change and ΔrS°m(T2) = ΔrS°m(298 K) + ΔrC°p, m ln (T2/298.15) for the entropy change, where ΔrS°m(298 K) = −80.85 J K−1 mol−1. The fit is far superior to the linear approximation. Nonetheless, a slight systematic deviation as T increases is observed. In order to improve the fit further would require adding a third (or more) term(s) in the expressions to account for the temperature dependence of ΔrC°p, m.
11.3.2 Response to pressure
Formally, the equilibrium constant does not depend on pressure

However, in reactions involving changes in volume, such as the example of methanol synthesis above or any other reaction that involves a change in the amount of substance of gas in units of moles, this does not mean that the equilibrium concentrations are independent of pressure. As an example, let us treat the gas-phase reaction N2O4  2NO2. We will derive an expression for the total pressure dependence of the equilibrium constant in terms of the fractional dissociation α, total pressure p, and the standard state pressure p°. The fractional dissociation is defined as α = ξ/a0. In the reaction chart below, ξ quantifies the amount of N2O4 that has reacted and a0 is the initial amount of substance. At constant pressure and temperature we can express the amount of substance in terms of the partial pressure. We will show that the fractional dissociation is a function of the total pressure even though the equilibrium constant itself is independent of p.
2NO2. We will derive an expression for the total pressure dependence of the equilibrium constant in terms of the fractional dissociation α, total pressure p, and the standard state pressure p°. The fractional dissociation is defined as α = ξ/a0. In the reaction chart below, ξ quantifies the amount of N2O4 that has reacted and a0 is the initial amount of substance. At constant pressure and temperature we can express the amount of substance in terms of the partial pressure. We will show that the fractional dissociation is a function of the total pressure even though the equilibrium constant itself is independent of p.
The reaction chart is

The last line is obtained by substituting ξ = αa0. With a0 as the initial partial pressure of NO2, then at equilibrium the partial pressure of NO2 is (1 − α)a0 and 2αa0 for N2O4. Therefore, the mole fractions at equilibrium are

The equilibrium constant is then

Solving for α yields

The equilibrium constant is constant with changing p because it is referenced to p°. As a consequence, when p increases α must decrease according to Eq. (11.23). Increasing p leads to decreasing α because, in accord with Le Châtelier’s principle, the reaction shifts to the left because this decreases the amount of substance of gas.
11.4 Equilibrium constants and associated calculations
11.4.1 Use of activities and concentrations
The Gibbs energy of reaction and the equilibrium constant are related by ΔrG°m = −RTln K. A large negative value of ΔrG°m favors products. Equivalently, for a large value of K, the products are favored. When ΔrG°m is positive or K«1, the reactants predominate. For ΔrG°m close to zero or K near one, the equilibrium mixture contains comparable concentrations of all species.
Equilibrium constants are defined in terms of activities/fugacities, not concentrations or partial pressures directly. The equilibrium constant written in terms of activities/fugacities is called the thermodynamic equilibrium constant. For the general reaction

the thermodynamic equilibrium constant is

Since activities are dimensionless, K is dimensionless. However, for systems behaving ideally such as dilute solutions, ideal gases and real gases at sufficiently low pressure, the approximation is often made of using concentration (or partial pressure) instead of activity. An equilibrium constant written in terms of concentrations Kc is sometimes referred to as the concentration equilibrium constant, and one written in terms of partial pressure Kp as the pressure equilibrium constant. By IUPAC recommended convention, all equilibrium constants are dimensionless quantities.
To write a thermodynamic equilibrium constant in terms of concentrations, we introduce the activity coefficients (or fugacity and fugacity coefficients for gases). The activity of species J is given the symbol aJ, and its activity coefficient is γJ. Activity is equal to the activity coefficient times concentration or pressure according to the choice of standard state. We will discuss activities, activity coefficients and their calculation in much greater detail in the chapters on solutions and mixtures. For now we only need to know that the activity coefficient quantifies the deviation from non-ideal behavior. As the system become more dilute and acts more ideally, the activity coefficient approaches one, γ → 1, and concentration (or partial pressure) can be used instead of activity. The concentration unit chosen for representing activity depends on the chosen standard state. Since activity is dimensionless, the concentration is divided by the concentration of the standard state to remove the units. Common choices of units and standard states are

While this is usually a good approximation for gases (at not too-high pressure or too-low temperature), this is a poor approximation for liquid solutions, especially for electrolytes at concentrations above ∼1 mM.
Volumes are temperature-dependent, masses are not. Therefore, molarity is temperature-dependent whereas molality is not. Historically, volumes were easier to measure than masses; thus, molarity was convenient with volumetric glassware. Now that we have electronic balances, mass is easy to measure and molality is preferred by many experimentalists. In terms of molalities the equilibrium constant is

The term Kγ accounts for deviations from ideality, while the term Kb is the thermodynamic equilibrium constant for an ideally behaved system. As the solution becomes more dilute γ → 1, Kγ → 1 and K → Kb. In this chapter, we want to emphasize the structure of concepts and calculations involving the equilibrium constant. Therefore, we will consider all systems to be behaving ideally unless we explicitly indicate otherwise. In other words, we assume γ = 1, Kγ → 1 and K → Kb. This will allow us to use concentrations in place of activities.
14.4.1.1 Directed practice
N2O4 dissociates according to the following equation

At 350 K, the equilibrium mixture contains 0.13 mol N2O4 and 0.34 mol NO2. The total pressure is 2 bar. Find the value of K at 350 K.
[Answer: 3.8]
14.4.1.2 Directed practice

At 400 K, K = 40. If 2.0 mol of H2 and I2 are mixed at a total constant pressure of 1 bar, what is the composition of the equilibrium mixture at 400 K?
[Answer: H2 = 0.5 mol, I2 = 0.5 mol, HI = 3.0 mol]
11.4.2 Heterogeneous/solution phase equilibria
An equilibrium involving two or more phases is a heterogeneous equilibrium, for example

Any species in its standard state has an activity of unity. Since their activities are one, they can be dropped from the equilibrium constant expression. A solid in its standard state has an activity of unity. A liquid in its standard state has an activity of unity. In a dilute solution, the solvent essentially acts like a substance in its standard state. Thus, the activities of solids and liquids can be dropped from equilibrium constant calculations for heterogeneous equilibria. The activities of solvents can be dropped from equilibrium constant calculations for solution phase equilibria as long as the solution is not too concentrated.
In the above examples, the solubility of AgCl(s) in H2O(l) at 25 °C is 1.9 × 10−4 g per 100 g of H2O. As long as AgCl is the only species being dissolved, the ions will never reach a concentration large enough to change the activity of water. Therefore, both the AgCl(s) and the H2O(l) can be dropped from the equilibrium constant expression. For glucose, on the other hand, the solubility at 80 °C is 81.5 mass% − the ‘solution’ is no longer a solution but a mixture in which the glucose is the majority component. In such a case the water can no longer be assumed to be acting ideally and the activity of water will differ from its concentration. The equilibrium constant expression for a liquid in contact with its vapor involves only the fugacity of the gas, as does the equilibrium expression for a solid that decomposes into gases.
11.4.2.1 Directed practice
Determine K for the following equilibrium at 298 K if the initial concentration of CH3NH2 is 0.10 mol dm−3 and the equilibrium concentration of OH− is 6.6 × 10−3 mol dm−3.
[Answer: K = 4.7 × 10−4]

11.4.3 Biochemical standard state
The physical chemistry standard state is defined in terms of molar units c° = 1 mol l−1 for all ions in solution and p° = 1 bar for all gases. In a biological setting, essentially all chemistry occurs in aqueous solution. It is impossible to have both H+ and OH− at 1 M concentration simultaneously. Therefore, in the biochemical standard state, the standard state concentration is c‘ = 1 mol l−1 for all ions other than H+ and OH−. The standard pressure is the same in both standard states. In the biochemical standard state, the aqueous phase is taken as neutral with pH 7, that is [H3O+] = [OH−] = 1.0 × 10−7 mol l−1. The difference between these standard states will only be apparent if we need to make comparisons between them or if we try to use values, for example, for standard state Gibbs energies obtained with one standard state in a system using the other standard state. The difference will only be important in reactions that involve either H+ or OH−. When looking up values in the literature you must always be aware of how the standard state is defined.
The choice of standard state does not change the relationship of Gibbs energy of reaction to equilibrium, ΔG = 0 still defines a system at equilibrium. Nor does the choice of standard state change the composition at equilibrium. The choice of standard state does change the value of the standard Gibbs energy of reaction. We denote the physical chemistry standard state value of the standard Gibbs energy of reaction in molar units as ΔrG°m. We denote the biochemical standard state value of the standard Gibbs energy of reaction in molar units as ΔrG′m.
We use the example of a simple solution-phase reaction A(aq) + B(aq)  Z(aq). In the physical chemical standard state, we can write the reaction quotient as
Z(aq). In the physical chemical standard state, we can write the reaction quotient as

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


