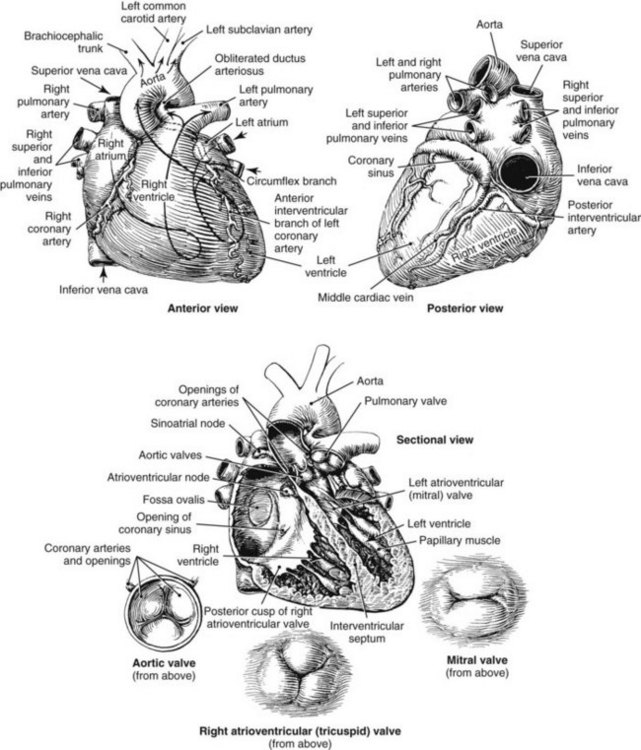
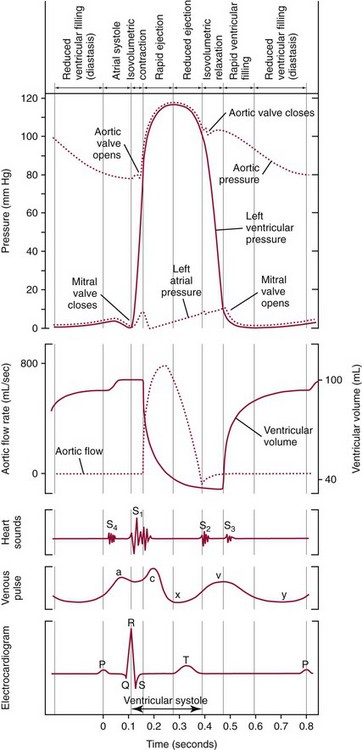
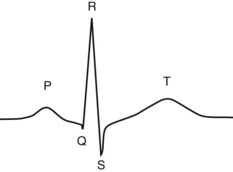
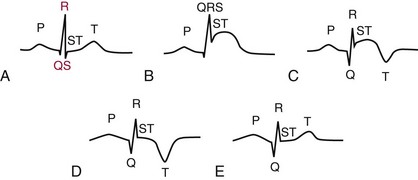
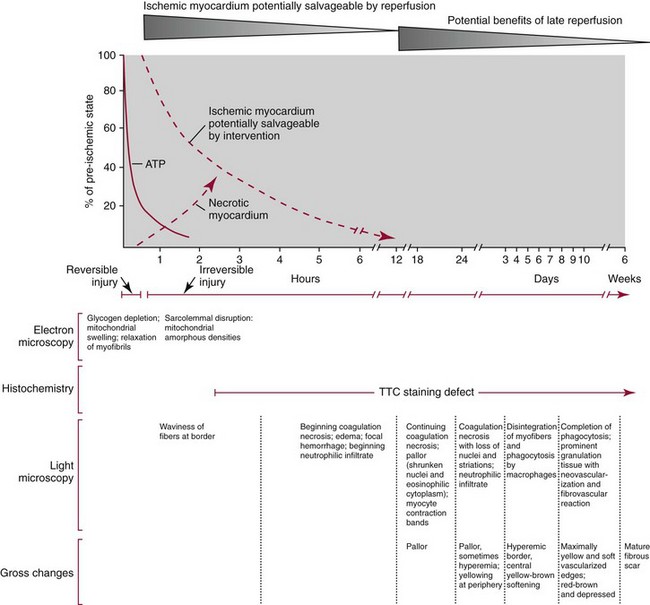
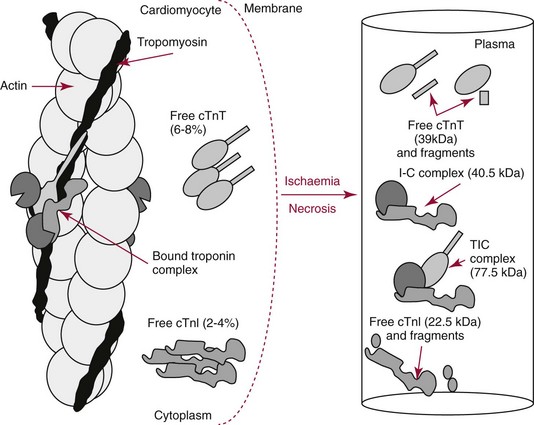
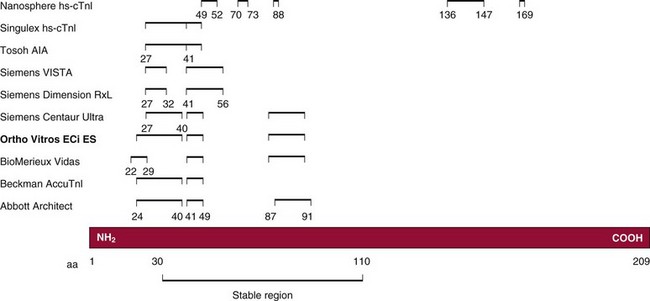
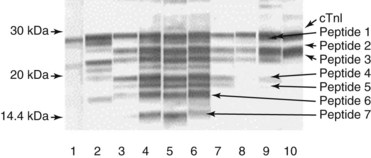
Chapter 47 Although the heart is an efficient and durable pump, a variety of pathologies are known to diminish cardiac function, possibly leading to a multiplicity of dysfunctional clinical states. Heart failure, which is increasing as we improve the treatment of acute ischemic heart disease, and acute ischemic heart disease itself are the most common cardiac diseases that rely on a biochemical diagnosis and thus will be the focus of this chapter.41 The term acute myocardial infarction (AMI) refers to a situation in which death of myocytes is due to an imbalance between myocardial oxygen supply and demand. When the blood supply to the heart is interrupted, “gross necrosis” of the myocardium results. In addition, a substantial number of cells die as the result of apoptosis. Such extensive damage is most often associated with a thrombotic occlusion superimposed on coronary atherosclerosis. Initially, it was thought that the population of myocytes was fixed; however, it is now believed that the migration of a variety of precursor stem cells has the potential at least to replace some of the damaged myocytes. It is now thought that the process of plaque rupture or erosion and thrombosis is one of the ways in which coronary atherosclerosis progresses, and that we recognize only more severe events.283,284 Total loss of coronary blood flow results in a clinical syndrome associated with what is known as ST segment elevation AMI (STE AMI). Partial loss of coronary perfusion if severe can lead to necrosis as well, which is generally less severe and is known as NSTEMI (non–ST elevation myocardial infarction). Other events of still lesser severity may be missed entirely or may be called angina, which can range from stable to unstable. Before the advent of coronary care units, treatment of AMI was directed toward allowing healing of the infarcted area. The concept that infarctions evolve over time and that their size can be moderated led to rethinking of this passive philosophy.319 We now know that re-establishment of perfusion reduces the extent of myocardial injury and is an important determinant of prognosis.330 Today the management of AMI suggested by most guidelines is aggressive and invasively oriented in the hope of reducing the extent of myocardial damage and thus improving prognosis.63A,67 In addition, prevention is finally being recognized as a key element in the long-term treatment of patients with atherosclerosis. The average human adult heart weighs approximately 325 g in men and 275 g in women and is 12 cm in length. The heart is a hollow muscular organ, shaped like a blunt cone, and is about the size of a human fist. It is located in the mediastinum, between the lower lobes of each lung, and rests upon the diaphragm. It is enclosed in a sac called the pericardium. The cardiac wall is composed of three layers: the epicardium, which is the outermost layer; a middle layer; and an inner layer, called the endocardium. The heart has four chambers. The two upper chambers are termed the right and left atria, and the two lower chambers are termed the right and left ventricles (Figure 47-1). Under normal circumstances, the atria are compliant structures, so that intracavitary pressure is low. When anatomy is normal, each atrium is connected to its ventricle through an atrioventricular (AV) valve, which opens and closes (see discussion later in this chapter). The valve on the left side is called the mitral valve and the one on the right side, the tricuspid valve. The right ventricle is banana shaped and pumps blood into the pulmonary artery through a tri-leaflet pulmonic valve. The left ventricle pumps blood into the aorta through a tri-leaflet aortic valve. The ventricles, especially the left ventricle, are thicker and less compliant in keeping with the need to generate higher pressures than the right ventricle, and intercavitary pressures are much higher than in the atria. Under normal conditions, the conduction or electrical system of the heart coordinates the sequential contraction of first the atria and then the ventricles. Given that they are connected, each side can affect the other. Under normal circumstances, the sequence of activation optimizes this interaction and thus the efficiency of cardiac function. The right and left coronary arteries originate from two of three cusps of the aortic valve. They provide blood flow and thus nutritive perfusion to the heart. The largest vessels are on the epicardium, and these can be accessed therapeutically fairly easily. Subsequent smaller branches divide to supply the remaining myocardium. The endocardium is the layer most susceptible to ischemia because its perfusion relies on the smallest vessels. A typical cardiac cycle consists of two intervals known as systole and diastole (Figure 47-2). During diastole, oxygenated blood returns from the lungs to the left atrium via the pulmonary veins, and deoxygenated blood returns from other parts of the body to fill the right atrium. During this period, the AV valves are open, allowing passive filling of the ventricle. At the end of diastole, the atria contract, forcing additional blood through the AV valves and into the respective ventricles. During systole, the ventricles contract. This closes the AV valves when ventricular pressure exceeds atrial pressure, and the pulmonary and aortic valves are opened when ventricular pressure exceeds pressure in the pulmonary arteries and/or the aorta, and blood flows into those conduits. During systole, a normal blood pressure in the aorta is typically 120 mm Hg; during diastole, it falls to about 70 mm Hg. At rest, the heart pumps between 60 and 80 times per minute. Stroke volume (i.e., the amount of blood expelled with each contraction) is roughly 50 mL, so cardiac output per minute is roughly 3 L. Typically, values are corrected for body surface area and are usually in the range of 2.5 to 3.6 L/min/m2. Measurements of cardiac output and ventricular filling pressures are the standards for assessing cardiac performance and function. Furthermore, therapeutic intervention in patients with heart disease often includes assessment of cardiac output and ventricular pressures. The cardiac cycle is tightly controlled by the cardiac conducting system, which initiates electrical impulses and carries them, via a specialized conducting system, to the myocardium. The surface electrocardiogram (ECG) records changes in potential and is a graphic tracing of the variations in electrical potential caused by excitation of the heart muscle and detected at the body surface.264 Clinically, the ECG is used to identify (1) anatomic, (2) metabolic, (3) ionic, and (4) hemodynamic changes in the heart. The clinical sensitivity and specificity of ECG abnormalities are influenced by a wide spectrum of physiologic and anatomic changes and by the clinical situation. Under normal circumstances, cardiac cycles are similar and each includes three major components (Figure 47-3): atrial depolarization (the P wave), ventricular depolarization (the QRS complex), and repolarization (the ST segment and T wave). Atrial depolarization, which is depicted by the P wave, produces atrial contraction. Ventricular depolarization, marked by the QRS complex, produces contraction of the ventricles. It is composed of as many as three deflections: (1) the Q wave, which when present is the first negative deflection; (2) the R wave, which is the first positive deflection; and (3) the S wave, which is a negative deflection following the R wave. On occasion, there is an R prime, which is a second positive deflection. Whether each of these occurs depends on the path of depolarization of the ventricles, as does the significance. Thus not every QRS complex will have discrete Q, R, and S waves. The ST segment and the T wave are produced by electrical recovery of the ventricles, and their mean electrical vector is under normal circumstances concordant (i.e., in roughly the same direction) with the mean QRS vector. CHF is a syndrome characterized by ineffective pumping of the heart leading to an accumulation of fluid in the lungs. Typically, it results from loss of cardiac tissue and subsequent function.88 Medically, it is defined as the pathophysiologic condition in which an abnormality of cardiac function is responsible for failure of the heart to pump sufficient blood to satisfy the requirements of metabolizing tissues. Encompassed in this definition are a wide spectrum of clinical conditions, ranging from (1) a primary impairment in pump function, such as might occur after a large AMI; (2) increased cardiac stiffness, which causes increased pressure in the heart, restricts filling, and increases hydrostatic pressures behind the area of reduced compliance; and (3) situations in which peripheral demand is excessive, resulting in what is known as high-output heart failure, which is defined as the inability of the heart to increase pumping sufficiently to meet the peripheral demands for blood. In the United States, CHF is the only cardiovascular disease with an increasing incidence. The National Heart, Lung, and Blood Institute estimates current prevalence at 4.9 million Americans with CHF, with an incidence of approximately 400,000 new cases each year.171 CHF is the leading cause of hospitalization in individuals 65 years of age and older. It has become clear that a larger component of heart failure is attributable to what has been called diastolic dysfunction, or heart failure (HF) with preserved ejection fraction (HFPEF). Therapeutic options for such patients are more limited than for those who have systolic abnormalities.313 Current prognosis is dependent on disease severity, but overall it is poor. Five-year mortality is approximately 10% in mild CHF, 20 to 30% in moderate CHF, and up to 80% in end-stage disease.205 These poor outcomes are not without substantial cost, estimated at $18.8 billion per year in the United States. The term acute coronary syndrome (ACS) encompasses patients who present with unstable ischemic heart disease.67 If they have ST segment elevation, they are called STEMI (Figure 47-4). Usually, but not always, these individuals will develop Q waves on their ECGs, hence the term Q-wave MI. If patients do not have STE but have biochemical criteria for cardiac injury, they are called NSTEMI, and most do not develop ECG Q waves. Those who have unstable ischemia and do not manifest necrosis are designated patients with unstable angina (UA). Most of these syndromes occur in response to an acute event in the coronary artery, when circulation to a region of the heart is obstructed for some reason. If the obstruction is high grade and persists, then necrosis usually ensues. Because necrosis is known to take some time to develop, it is apparent that opening the blocked coronary artery in a timely fashion can often prevent some of the death of myocardial tissue. This is clearly the case with STEMI. With non-STEMI [American Heart Association (AHA)/American College of Cardiology (ACC) guidelines], early but not immediate intervention is advocated, because most often the infarct-related coronary artery is not totally occluded, and thus immediate intervention is less necessary. These syndromes are usually but not always associated with chest discomfort (see discussion later in this chapter).286 The major cause of ACS is atherosclerosis, which contributes to significant narrowing of the artery lumen and a tendency for plaque disruption and thrombus formation.67,283,284 Myocardial ischemia and infarction are usually segmental diseases. In up to 90% of patients with these diseases, focal occlusion of only one of the three large coronary vessels or branches occurs. The resulting impaired contractile performance of that segment occurs within seconds and is initially restricted to the affected segment(s). Myocardial ischemia and subsequent infarction usually begin in the endocardium and spread toward the epicardium.319 The extent of myocardial injury reflects (1) the extent of occlusion, (2) the needs of the area deprived of perfusion, and (3) the duration of the imbalance in coronary supply. Irreversible cardiac injury consistently occurs in animals when the occlusion is complete for at least 15 to 20 minutes. Most damage occurs within the first 2 to 3 hours. Restoration of flow within the first 60 to 90 minutes evokes maximal salvage of tissue, but benefits of increased survival are possible up to 4 to 6 hours. In some situations, the restoration of coronary perfusion even later is of benefit.330 The percentage of tissue at risk of necrosis (infarct size) depends on the amount of antegrade flow, the existing collateral flow, which is highly variable and difficult to predict, and the metabolic needs of the tissue.246,328,366 In almost all instances, the left ventricle is affected by AMI. However, with right coronary and/or circumflex occlusion, the right ventricle can also be involved, and there is a clinical syndrome in which damage to the right ventricle predominates and is the major determinant of hemodynamics. Coronary thrombi will undergo spontaneous lysis, even if untreated, in about 50% of cases within 10 days. However, for patients with STE AMI, opening the vessel earlier with clot-dissolving agents (thrombolysis) and/or percutaneous intervention can often save myocardium and lives (Figure 47-5). At present, immediate percutaneous intervention (PCI) with stenting is the preferred therapy for STE AMI. However, many hospitals cannot or do not offer urgent PCI 24 hours a day, 365 days per year. Thus clot-dissolving medications still play a major role in the treatment of these patients. In addition, it is now apparent that urgent but not necessarily immediate invasive revascularization benefits those with NSTEMI.67 These individuals usually have only partial coronary occlusion and smaller amounts of cardiac damage acutely. However, untreated, repetitive episodes often eventually damage larger amounts of myocardium, leading to increased morbidity and mortality over time. Treatments such as newer anticoagulants and antiplatelet and anti-inflammatory agents, in conjunction with coronary revascularization, save lives in this group. The prognosis for patients with ischemia but without necrosis is far better. Some studies based on biomarkers would suggest that in patients with no troponin elevation, interventional therapies may be harmful.395 A major determinant of mortality and morbidity is the amount of myocardial damage that occurs. With STE AMI, most damage is acute, whereas with NSTEMI, damage may evolve as the result of repetitive events over many months; thus, interrupting the process improves survival. In many patients with AMI, no precipitating factor can be identified. Studies have noted the following patient activities at the onset of AMI: (1) heavy physical exertion, 13%; (2) modest or usual exertion, 18%; (3) surgical procedure, 6%; (4) rest, 51%; and (5) sleep, 8%. Exertion before infarction is somewhat more common among patients without preexisting angina than in those who have a history of angina.342 Causes of infarction other than acute atherothrombotic coronary occlusion have been identified. For example, prolonged vasospasm can induce infarction, and spontaneous dissection is becoming more commonly appreciated, especially in pregnant females.67 In addition, it is now clear that some patients, particularly women, can have acute infarction with normal-appearing angiographic coronary arteries.247 Other conditions (Box 47-1) can also cause the death of cardiomyocytes, leading to a biochemical signal (such as increased circulating concentrations of cardiac troponins) of myocyte damage, but these conditions should not be confused with myocardial infarction.176 Pulmonary embolism (PE) is another common cause of biochemical elevations that is caused by right ventricular damage related to acute increases in wall stress and reduced subendocardial perfusion.132,170,216 There is a pronounced periodicity for the time of onset of STE AMI.68,115,342 Often an AMI occurs in the morning hours soon after arising; this is a period of (1) increasing adrenergic activity, (2) increased plasma fibrinogen levels, (3) increased inhibition of fibrinolysis, and (4) increased platelet adhesiveness. Studies have demonstrated that the early morning peak in MI parallels the peak incidence of death from ischemic heart disease, which occurs at about 8 AM to 9 AM. A second peak has been noted at approximately 5 PM. Diurnal differences affect many physiologic and biochemical parameters; the early morning hours are associated with rises in plasma catecholamines and cortisol and increases in platelet aggregability. Tissue plasminogen activator activity is low and plasminogen activator inhibitor activity is high during the early morning hours. Thus it is possible that some cyclic aspects of combined vasospastic, prothrombotic, and fibrinolytic factors, in the setting of preexisting atherosclerosis, lead to AMI. NSTEMI does not exhibit this diurnal pattern. STE and non-STE infarctions have distinctly different short-term prognoses. STE AMI is associated with higher early and in-hospital mortality. It is said that mortality associated with STE AMI can occur up to 6 months post event, but the vast majority (at least two thirds) occur during the first 30 or 40 days. It is this risk that coronary recanalization seems to benefit. NSTE AMI is associated with lower acute mortality and complication rates but a longer period of vulnerability to reinfarction and death. As a result, 1- to 2-year survival rates are similar to those for STEMI.67 This is why intervention has been so effective in this group. The clinical history remains of substantial value in establishing a diagnosis.330 A prodromal history of angina can be elicited in 40 to 50% of patients with AMI. Among patients with AMI who present with prodromal symptoms, approximately one third have had symptoms from 1 to 4 weeks before hospitalization; in the remaining two thirds, symptoms predate admission by a week or less, with one third of patients having had symptoms for 24 hours or less. Figure 47-5 shows the temporal sequence of early biochemical, histochemical, and histologic findings after the onset of AMI. On gross pathologic examination, AMI can be divided into subendocardial (nontransmural) infarctions and transmural infarctions.11 In the former, necrosis involves the endocardium, the intramural myocardium, or both without extending all the way through the ventricular wall to the epicardium. In the latter, myocardial necrosis involves the full thickness of the ventricular wall. The histologic pattern of necrosis may differ: contraction band injury occurs almost twice as often in nontransmural infarctions as in transmural infarctions. Unfortunately, the pathologic changes correlate poorly with clinical, ECG, and biochemical markers of necrosis, which is why those terms are no longer used clinically. Statistically, patients are more apt to have STE MI Q waves on the ECG and larger biochemical signals when the infarction is transmural pathologically. Although it was previously believed that no light microscopic changes could be seen in infarcted myocardium until 8 hours after interruption of blood flow, in some infarcts a pattern of wavy myocardial fibers may be seen 1 to 3 hours after onset, especially at the periphery of the infarct.11 After 8 hours, edema of the interstitium becomes evident, as do increased fatty deposits in the muscle fibers, along with infiltration of neutrophilic polymorphonuclear leukocytes and red blood cells. Gross alterations of the myocardium are difficult to identify until at least 6 to 12 hours following the onset of necrosis.11 However, several histochemical approaches have been used to identify zones of necrosis that can be observed after only 2 to 3 hours. Initially, the myocardium in the affected region may appear pale and slightly swollen. By 18 to 36 hours after onset of the infarct, the myocardium is tan or reddish purple (because of trapped erythrocytes). These changes persist for approximately 48 hours; the infarct then turns gray, and fine yellow lines, secondary to neutrophilic infiltration, appear at its periphery. This zone gradually widens and during the next few days extends throughout the infarct. Intrinsic to modern day understanding of ischemic heart disease and to the intense interest in the development of markers of inflammation is the concept that atherosclerosis is a chronic inflammatory disease. The concept is that some event damages the endothelium of blood vessels, which facilitates the egress of lipid into the subendothelial space. Putative injurious stimuli include turbulent flow in a blood vessel, which could occur for example because of hypertension or a noxious metabolite from a lipid fraction. This damage tends to occur at branch points of blood vessels. Regardless of the initial stimulus, once damaged, low-density lipoprotein (LDL) can cross into the vessel wall more easily in a nicotinamide adenine dinucleotide phosphate (NADPH) oxidase–mediated fashion. Whether minimal oxidation facilitates that egress or whether it occurs once the LDL is within the vessel wall is unclear, but a minimal degree of oxidation once in the vessel wall facilitates the egress of smooth muscle cells from the media of the vessel and macrophages that ingest cholesterol, hence the rationale for the measurement of oxidized lipids in blood. The process of atherosclerosis progresses slowly with involvement of lymphocytes, monocytes, macrophages, and smooth muscle cells. The dynamic within a given plaque may vary, but there clearly is an inflammatory milieu, in part mediated by substances such as CD40 ligand, which can be measured directly or indirectly as C-reactive protein (CRP). Interleukins-1, -6, -8, and -18 also participate to various extents as part of this chronic inflammatory process. This process involves adherence of white blood cells to the damaged endothelial surface with subsequent degranulation and elaboration of myeloperoxidase. A procoagulant component is due predominantly to the presence of tissue factor, which is localized immediately under the cap of the plaque. Intermittent instability is noted because of inflammatory products within the plaque that release chemicals, such as metalloproteinases. Initially the plaque expands by stretching the adventitia through a process of small ruptures with release of procoagulant and proinflammatory materials and then remodeling over time as anti-inflammatory and anticoagulant and thrombolytic substances are elaborated. This process of stretching the adventitia preserves the lumen such that by the time luminal encroachment occurs, there is a very large plaque burden.136 A categorization of plaques has been proposed to facilitate identification of those at risk of rupture that could lead to an acute event. It is acknowledged that the propensity for a plaque to rupture probably reflects a systemic predilection rather than a local one. Thus for a given patient at risk, there likely are many plaques that are metabolically at risk of rupture at any given time.283,284 High-risk plaques have 1. An active inflammatory environment that not only may be intrinsic but may be stimulated additionally by systemic infection. 2. A thin fibrous cap on the endothelial surface with a large lipid core that is filled with procoagulant substances, predominantly tissue factor. 3. Endothelial denudation and fissuring caused by the elaboration of metalloproteinases. 4. Local high shear stress, usually because they are severe, at branch points in the vessel. The diagnosis of AMI established by the World Health Organization in 1986 included biomarkers as an integral part of the disorder and required that at least two of the following criteria be met: (1) a history of chest pain, (2) evolutionary changes on the ECG, and/or (3) elevations of serial cardiac markers to a level two times the normal value. However, over time, it became rare for a diagnosis of AMI to be made in the absence of biochemical evidence of myocardial injury. A 2000 European Society of Cardiology/American College of Cardiology (ESC/ACC) consensus conference66 updated in 2007 (Global Task Force)372 codified the role of markers by advocating that the diagnosis should be regarded as evidence of myocardial injury based on markers of cardiac damage in the appropriate clinical situation (Box 47-2).372 The criteria for diagnosis of an established MI are listed in Box 47-3. The guidelines thus recognized the reality that neither the clinical presentation nor the ECG had adequate sensitivity and specificity. This guideline does not suggest that all elevations of these biomarkers should elicit a diagnosis of AMI—only those associated with appropriate clinical and ECG findings (see discussion later in this chapter). When elevations that are not caused by acute ischemia occur, the clinician is obligated to search for another cause for the elevation.176,181,401,406 The criteria suggested for use with these markers by the Biochemistry Panel of the ESC/ACC Committee are listed in Box 47-4.181 In the 2007 revision of the guidelines, several types of AMI were recognized, including the spontaneous type, which is associated with plaque rupture or erosion, and the type associated with fixed or transient coronary abnormalities but not thrombotic occlusion. These are discussed in greater detail in the following paragraphs. It is also recognized that one can have a classic AMI and succumb before markers are obtained or become elevated, and cardiac injury can occur in association with cardiac procedures.372 In addition, criteria for use with coronary interventions and bypass surgery were suggested. At one time, the initial ECG was thought to be diagnostic of AMI in about 50% of patients.179 As the frequency of STE AMI has diminished and the diagnosis has been made with greater and greater sensitivity, this percentage has been greatly reduced. Serial tracings are helpful for STE AMI but not for what is now almost 70% of AMIs that are known as non-STE (NSTE) AMIs. The classic ECG changes of an STE AMI are ST segment elevation, which often evolves to the development of Q waves if intervention is not provided (see Figure 47-4). Pericarditis, some normal variants, and transient causes that may result in myocardial injury such as myocarditis are well described and on occasion can mimic the changes of AMI. Most NSTE AMIs present as ST segment depression, with or without T-wave changes; as T-wave changes alone; or on occasion in the absence of any ECG findings. Those with ST segment change have a substantially worse prognosis.372 The contractile proteins of the myofibril include the three troponin regulatory proteins (Figure 47-6).85,196,244 The troponins are a complex of three protein subunits: troponin C (the calcium-binding component), troponin I (the inhibitory component), and troponin T (the tropomyosin-binding component). The subunits exist in a number of isoforms. The distribution of these isoforms varies between cardiac muscle and slow and fast twitch skeletal muscle. Only two major isoforms of troponin C are found in human heart and skeletal muscle. These are characteristic of slow and fast twitch skeletal muscle. The heart isoform is identical to the slow twitch skeletal muscle isoform, thus the reason why cTnC was never developed as a cardiac specific biomarker. Isoforms of cardiac-specific troponin T (cTnT) and cardiac-specific troponin I (cTnI) also have been identified and are the products of unique genes.62,63,94,194 Troponin is localized primarily in the myofibrils (94 to 97%), with a smaller cytoplasmic fraction (3 to 6%).122 Some experts in the field think that 100% of cTn is myofibril bound and that the “cytoplasmic fraction” represents a more easily mobilizable fraction, rather than representing a different cellular localization. cTnI and cTnT have different amino acid sequences from the skeletal isoforms and are encoded by unique genes. Human cTnI has an additional 31-amino-acid residue on the amino terminal end compared with skeletal muscle TnI, giving it complete cardiac specificity (Figure 47-7). Only one isoform of cTnl has been identified. cTnI has never been shown to be expressed in normal, regenerating, or diseased human or animal skeletal muscle.62 cTnT is encoded for by a different gene than the one that encodes for skeletal muscle isoforms. An 11 amino acid amino-terminal residue gives this marker unique cardiac specificity. However, during human fetal development, in regenerating rat skeletal muscle, and in diseased human skeletal muscle, small amounts of cTnT are expressed as one of four identified isoforms in skeletal muscle.10,63,335 In humans, cTnT isoform expression has been demonstrated in skeletal muscle specimens obtained from patients with muscular dystrophy, polymyositis, dermatomyositis, and end-stage renal disease.63,255,298,320,371 Thus, care is necessary to choose antibody pairs for the cTnT assay that do not detect these re-expressed isoforms.130,217 A substantial body of evidence shows that following myocardial injury or because of genetic disposition, multiple forms of cTn are elaborated both in tissue and in blood (Figure 47-8).193,194,406 These include the following: T-I-C ternary complex, IC binary complex, and free I; multiple modification of these three forms can occur, involving oxidation, reduction, phosphorylation, and dephosphorylation, as well as both C- and N-terminal degradation. Depending on the selection of antibodies used to detect cTnI, different antibody configurations can lead to a substantially different recognition pattern (Figure 47-9).85 The conclusions derived from these observations are that assays need to be developed in which the antibodies recognize epitopes in the stable region of cTnI and, ideally, demonstrate an equimolar response to the different cTnI forms that circulate in the blood. Cummins and coworkers were the first to develop an RIA to measure cTnI, using polyclonal anti-cTnI antibodies.94 The first monoclonal enzyme-linked immunosorbent assay (ELISA), an anti-cTnI antibody–based immunoassay, was described by Bodor and Ladenson.61 Numerous manufacturers have now described the development of monoclonal antibody–based diagnostic immunoassays for the measurement of cTnI in serum.14,17,18,22,28,29,34,37,85,87,90,161,195 As shown in Table 47-1, numerous assays have been approved by the U.S. Food and Drug Administration (FDA) for patient testing within the United States on central laboratory and POC testing platforms. In addition to these quantitative assays, several assays have been cleared by the FDA for the qualitative determination of cTnI. Further development has led to the design of research (prototype) high-sensitivity cTn (hs-cTn) I and T assays. In practice, two obstacles limit the ease of switching from one assay to another in clinical practice or research. First, no primary reference cTnI material is currently available for manufacturers to use in standardizing cTnI assays. Second, assay concentrations fail to be consistent because cTnI circulate in its various forms and the different antibodies use in the available assays recognize different epitopes of cTnI. TABLE 47-1 The cTnI standardization subcommittee of the American Association for Clinical Chemistry (AACC) in collaboration with the National Institute of Standards and Technology (NIST) developed a cTnI reference material (SRM #2921) that is a TnC-cTnI-cTnT complex purified from human heart under nondenaturing conditions.74 A cTnI value was assigned by a combination of reversed-phase liquid chromatography with ultraviolet detection and amino acid analysis. However, because this material demonstrated commutability with only 50% of current cTnI assays, it is of limited value for assay harmonization of current assays and cannot be used as a common calibrator. Although standardization of assays remains elusive, differences of cTnI concentrations reported by studied assays has been narrowed from a 20-fold difference to a twofold to threefold difference using the SRM 2921. Further, this material allows for traceability to a common reference material. It appears that the only way to achieve complete standardization for cTnI would be for manufacturers to agree to use the same antibodies showing similar specificity for the cTnI molecule; this would also overcome matrix effects with a serum-based common reference material for calibration. Several adaptations of the Roche cTnT immunoassay have been described over the years.383,405 The current FDA-cleared assay available worldwide involves two monoclonal, anti–cardiac troponin T antibodies. Skeletal muscle TnT is no longer a potential interferant, as was found in the first-generation ELISA cTnT assay. In contrast to cTnI, no standardization bias exists for cTnT, because the same antibodies (M11, M7) are used in the central laboratory and in POC assay systems. In 2001 and 2004, the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) Committee on Standardization of Markers of Cardiac Damage (C-SMCD) recommended quality specifications for cTn assays.300,301 These specifications were intended for use by the manufacturers of commercial assays and by clinical laboratories utilizing cTn assays. The overall goal was to attempt to establish uniform criteria so that all assays could be evaluated objectively for their analytical qualities and clinical performance. Both analytical and preanalytical factors were addressed and are in the process of being updated with an expected publication date of 2012. An adequate description of the analytical principles, method design, and assay components should include the following: (1) antibody specificity and epitope locations identified need to be delineated; (2) epitopes located on the stable part of the cTnI molecule should be a priority; (3) assays need to clarify whether different cTnI forms (e.g., binary vs. ternary complex) are recognized in an equimolar fashion by the antibodies used; (4) specific relative responses need to be described for each of the cTnI forms (free cTnI, the 1-C binary complex, the T-I-C ternary complex, and oxidized, reduced, and phosphorylated isoforms of the three cTnI forms); (5) the effects of different anticoagulants on binding of cTnI need to be addressed; and (6) the source of material used to calibrate cTn assays, specifically for cTnI, should be reported (Box 47-5). Advancements in cTn assay technology have created a conundrum for clinicians and laboratory scientists, who must determine which assays are best for optimal patient care. Unfortunately, few resources are available to guide the medical and scientific communities in this regard. International guidelines have defined an increased cTn above the 99th percentile limit as an abnormal result, as described in the clinical section.17,273,372 What is lacking, unfortunately, is an approach to define the 99th percentile limit across the heterogeneity of assays. In spite of evidence-based literature demonstrating that cTn concentrations tend to increase in individuals older than 60 years, likely because of unrecognized comorbidities, 99th percentile reference limits are often determined across wide age ranges using subjects as old as 80 years (convenience samples).14,21,116 Further frustrating the problem of selecting relevant reference subjects is the fact that in clinically defined normal individuals without known cardiovascular disease, increased cTn concentrations are indicative of a significantly higher risk of death. Given such problems, most laboratories (1) accept the manufacturer’s reference limit from the FDA-cleared package insert; (2) perform an underpowered normal range study to establish a reference limit; or (3) accept a cutoff value published in the literature. Consensus guidelines from the Global Task Force for the Universal Definition of Myocardial Infarction and the National Academy of Clinical Biochemistry (NACB) plus the updated American College of Cardiology/American Heart Association and Epidemiology133,240 guidelines have recommended that, in patients who present with ischemic symptoms, at least 1 cTn concentration higher than the 99th percentile value during the first 24 hours after onset of symptoms indicates myocardial necrosis. If this elevation occurs in a clinical situation consistent with myocardial infarction (MI), that diagnosis should be made (see Box 47-2). It is recommended that cTn assays with appropriate quality control and optimal total imprecision [coefficient of variation (CV) ≤10%] at the 99th percentile limit are preferred.86,301 Better imprecision at low cTn concentrations appears to improve the value of cTn as a diagnostic and risk indicator. Use of cTn assays with intermediate imprecision (10 to 20% CV) at the 99th percentile, however, does not lead to significant patient misclassification when serial cTn results are interpreted. A challenge that arises as the FDA clears improved cTn assays with higher analytical sensitivity for use in laboratory practice is determining how these new assays compare with the older assays. Diagnostic sensitivities using specimens collected at presentation for detection of myocardial infarction (MI) have improved from 15 to 35% for early cTn assays to 50 to 75% for contemporary assays (Table 47-2). Unfortunately, definitive data about how long it takes to totally exclude AMI are still lacking. Over the past 10 years, the science of reagent and technology formulation of cTn assays has allowed measurement of this biomarker with greatly improved precision at concentrations approaching the 99th percentile limit of an assay. The goal is to better define the clinical playing field for assays used to assist the diagnosis of MI and to better stratify patients by risk of adverse events. To summarize these discussions, the optimal goals that the FDA would like to achieve include the following: (1) to transition from the current practice of approving assays based on receiver operating characteristic (ROC)-optimized cutoffs to using the 99th percentile value; (2) to ensure that cTn assays are accurate enough at the 99th percentile values for clinical use; and (3) to define the effects of implementing the 99th percentile value in clinical practice on minimizing false-negative and false-positive findings. Understanding what is truly normal for cTn, once high-sensitivity assays are incorporated into clinical practice, will be a major step forward in the cardiac biomarker field. The authors advocate for worldwide acceptance of the 99th percentile cTn value as the MI diagnostic cutoff—not ROC curve–derived cTn cutoff values. TABLE 47-2 Diagnostic Accuracy of Current cTn Assays 99th Percentile at Presentation of Patient to Hospital
Cardiac Function
Basic Anatomy
Physiology
Cardiac Conducting System
Cardiac Disease
Congestive Heart Failure
Epidemiology
Acute Coronary Syndrome
Precipitating Factors
Chronobiology
Prognosis
Clinical History
Myocardial Changes Following Acute Myocardial Infarction
Histologic (Light Microscopic) Changes in Myocardium
Gross Changes in Myocardium
Development and Progression of Atherosclerosis229
Diagnosis of Acute Myocardial Infarction
ECG Findings
Biomarkers In Acute Coronary Syndrome
Biochemistry
Immunoassays
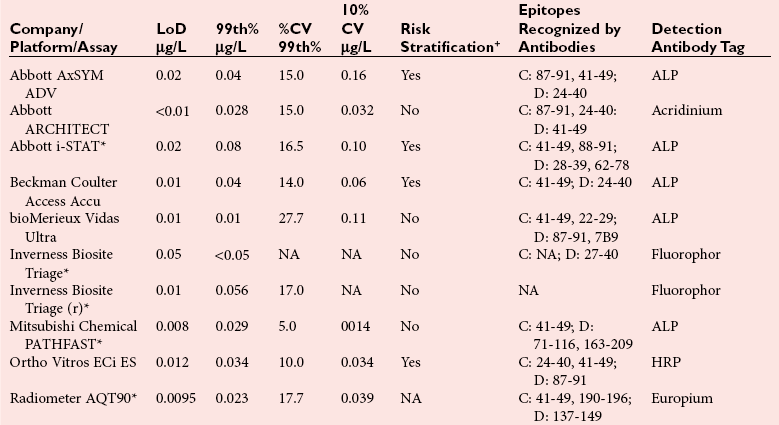
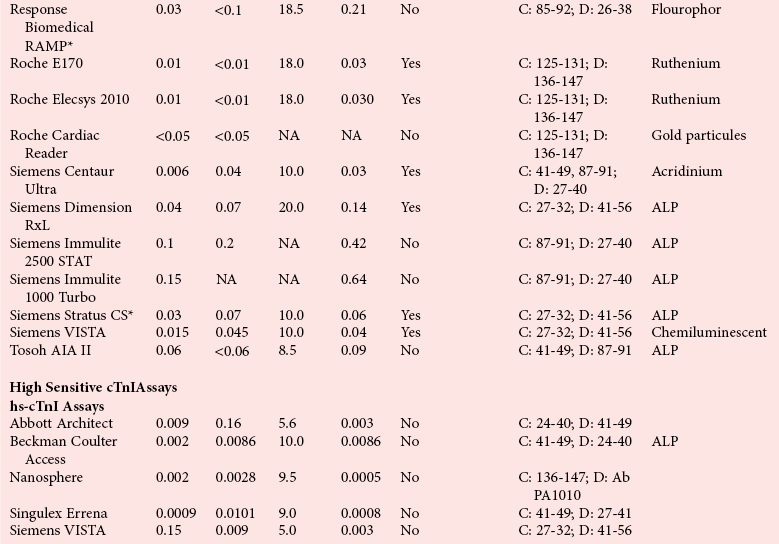
Assay Specifications
Defining Normal Reference Limits for Cardiac Troponin
Sensitivity
Specificity
2003 Collinson, Roche Elecsys cTnT
82%
60%
2004 Eggers, Siemens Dimension cTnI
78%
NA
2006 Apple, Siemens Stratus CS cTnI
68%
82%
2007 Apple, Ortho-Clinical Diagnostics ECi cTnI ES
83%
79%
2007 Apple, bioMerieux VIDAS cTnI
88%
79%
2008 Apple, Siemens Centaur Ultra cTnI
74%
83%
2009 Reichlin, Abbott ARCHITECT cTnI
86%
92%
2009 Reichlin, Roche Elecsys cTnI
84%
94%
2009 Reichlin, Roche Elecsys hs-cTnT
95%
80%
2011 Apple, Mitsubishi PATHFAST
73%
92.7%
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Cardiac Function